PROTEAN CR: Proteomics Toolkit for Ensemble Analysis in Cancer Research
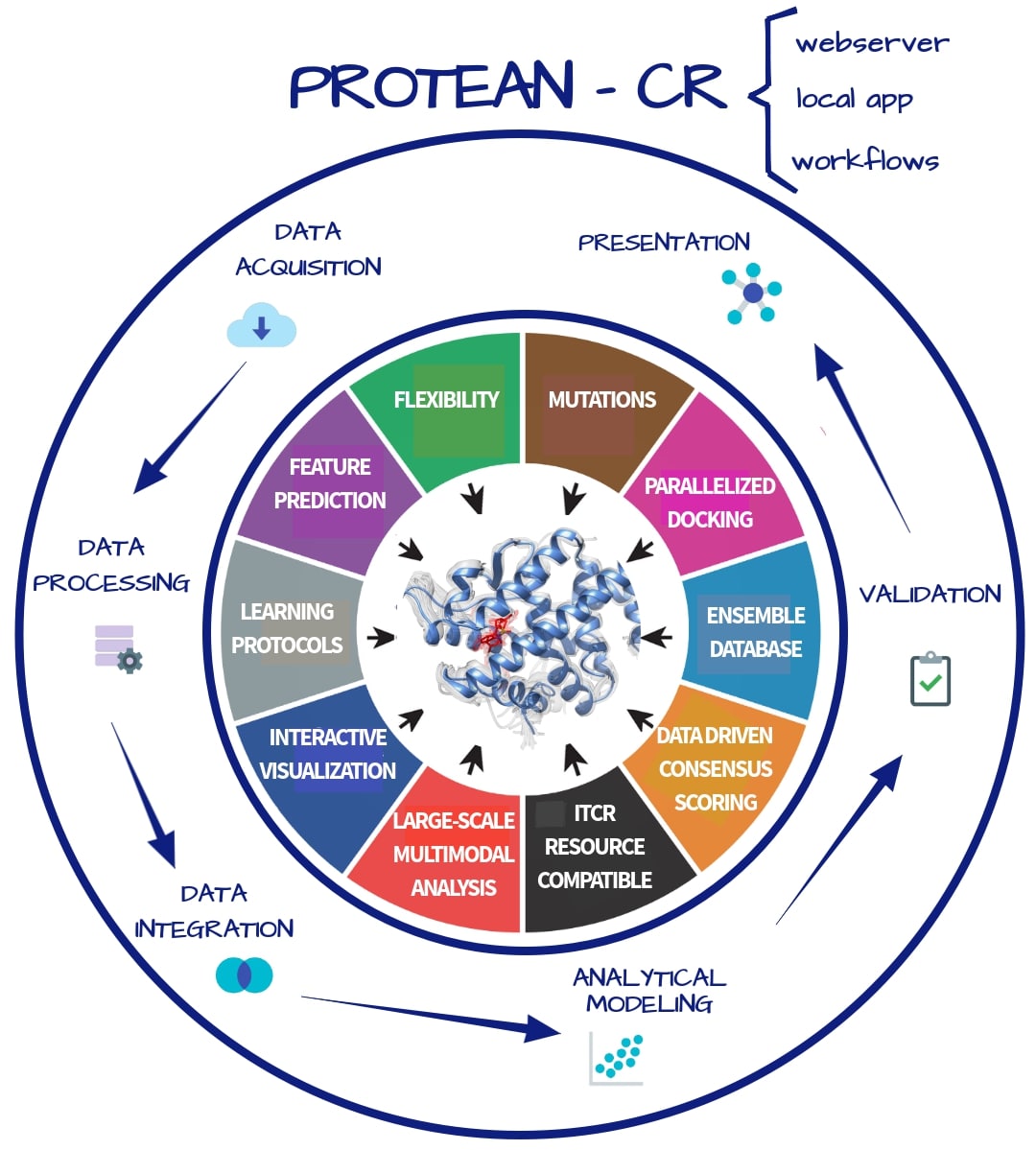
Understanding protein–ligand molecular interactions is fundamental to understanding the role of proteins in complex diseases such as cancer. For instance, there is growing interest in predicting the binding modes of peptide-based ligands (e.g., cyclic and phosphorylated peptides) to inhibit or induce targeted degradation of high-profile cancer targets. Another promising example is the identification of tumor-associated antigens for cancer immunotherapy applications. Both examples involve very specific molecular interactions, provide opportunities for computer-aided design of better cancer treatments, and highlight the need for structural analyses in cancer research. They also require new methods that account for the flexibility and variability of the protein receptors involved in these molecular interactions. The objective of this project is to develop an integrated approach to the structural modeling and analysis of protein–ligand interactions in cancer research that will be implemented in the proteomics toolkit PROTEAN-CR. The proposed toolkit will adopt a data-science approach to the problem by introducing approaches for data acquisition and aggregation, as well as algorithmic advances for handling receptor flexibility and for modeling driver mutations, drug-resistance polymorphisms, and post-translational modifications. PROTEAN-CR will streamline running structural analyses at scale while providing meaningful data analytics. The long-term goal of our research is to fully integrate three-dimensional structural information about proteins and ligands and structural analysis into cancer research. This project is intended to target a wide range of users, from experimentalists with little to no programming experience, to advanced users who are comfortable scripting large-scale analyses and integrating the toolkit with their own computational pipeline. The central hypothesis is that a unified data-science-inspired approach can be used to address major challenges in structural analysis of protein–ligand interactions in cancer research at scale. The first aim will incorporate protein flexibility in docking studies for cancer research. Specific workflows will be used to generate ensembles of protein conformations (receptor flexibility) and innovative machine learning methods will be implemented aiming at a better scoring of protein–ligand complexes. The second aim will focus on including cancer variability into structural analysis. We aim to fill the gap that exists between available data on cancer variants and the structural analysis of ensembles of tumor-associated mutations and protein modifications. Finally, the third aim will focus on customization, interpretability and scalability, where user-friendly methods will be deployed to manage ensembles of protein-ligand complexes. PROTEAN-CR will be developed focusing on specific cancer-related projects, and with a broad network of collaborators, enabling the design, implementation and evolution of the tool according to the needs of the cancer research community. More information available at https://reporter.nih.gov/search/DZaxB9c7-kWkwmA8MnbGVg/project-details/10188196
This work has been supported by grant NCI 1U01CA258512-01.
Related Publications
- J. K. Slone, M. Zhang, P. Jiang, A. Montoya, E. Bontekoe, B. N. Rausseo, A. Reuben, and L. E. Kavraki, “STAG-LLM: Predicting TCR-pHLA binding with protein language models and computationally
generated 3D structures,” Computational and Structural Biotechnology Journal, vol. 27, pp. 3885–3896, Sep. 2025.
BibTeX
@article{slone-stagllm2025, title = {STAG-LLM: Predicting TCR-pHLA binding with protein language models and computationally generated 3D structures}, journal = {Computational and Structural Biotechnology Journal}, volume = {27}, pages = {3885-3896}, year = {2025}, month = sep, issn = {2001-0370}, doi = {10.1016/j.csbj.2025.09.004}, url = {https://www.sciencedirect.com/science/article/pii/S2001037025003642}, author = {Slone, Jared K. and Zhang, Minying and Jiang, Peixin and Montoya, Amanda and Bontekoe, Emily and Rausseo, Barbara Nassif and Reuben, Alexandre and Kavraki, Lydia E.}, keywords = {Structural bioinformatics, Proteomics, Immunology, Protein language model, Geometric deep learning, TCR, HLA}, abstract = {Background: Strong binding between T cell receptors (TCRs) and peptide–HLA (pHLA) complexes is important for triggering the adaptive immune response. Binding specificity prediction, identifying which TCRs will bind strongly to which pHLAs, can serve as a first step in designing personalized immunotherapy treatments. Existing machine learning (ML) methods to predict binding specificity rely primarily on the amino acid sequences of TCRs and pHLAs to make predictions. However, incorporating the 3D structure and geometry of the TCR-pHLA complex as an additional data modality alongside protein sequence offers a promising approach to improving ML methods for predicting TCR-pHLA binding specificity. Modern computational modeling tools present unprecedented opportunities to incorporate structure data into ML pipelines. We utilize such computational tools to incorporate 3D data into this work. Results: We present STAG-LLM, a multimodal ML model for predicting TCR-pHLA binding specificity that leverages sequence data and computationally generated 3D protein structures. We show that by combining a protein language model with a geometric deep learning architecture, our method outperforms existing methods even when trained on 3x smaller datasets. To further validate our model, we conduct in vitro alanine scanning experiments for four peptides and demonstrate a correlation with the attention weights learned by our model and in vitro results. We also seek to address three key challenges that arise from using computationally generated 3D structures in ML pipelines: increased inference costs arising from the need to generate 3D structures, limited training data, and robustness to noise in the generated structures. Conclusions: STAG-LLM shows tremendous potential for structure-based TCR-pHLA binding prediction methods, offering a foundation for further advancements in using modeled 3D structures to solve problems in immunology and proteomics. We anticipate that the usefulness of STAG-LLM and similar tools will increase in coming years as both protein structure prediction models and large language models continue to advance.} }DetailsAbstract
Background: Strong binding between T cell receptors (TCRs) and peptide–HLA (pHLA) complexes is important for triggering the adaptive immune response. Binding specificity prediction, identifying which TCRs will bind strongly to which pHLAs, can serve as a first step in designing personalized immunotherapy treatments. Existing machine learning (ML) methods to predict binding specificity rely primarily on the amino acid sequences of TCRs and pHLAs to make predictions. However, incorporating the 3D structure and geometry of the TCR-pHLA complex as an additional data modality alongside protein sequence offers a promising approach to improving ML methods for predicting TCR-pHLA binding specificity. Modern computational modeling tools present unprecedented opportunities to incorporate structure data into ML pipelines. We utilize such computational tools to incorporate 3D data into this work. Results: We present STAG-LLM, a multimodal ML model for predicting TCR-pHLA binding specificity that leverages sequence data and computationally generated 3D protein structures. We show that by combining a protein language model with a geometric deep learning architecture, our method outperforms existing methods even when trained on 3x smaller datasets. To further validate our model, we conduct in vitro alanine scanning experiments for four peptides and demonstrate a correlation with the attention weights learned by our model and in vitro results. We also seek to address three key challenges that arise from using computationally generated 3D structures in ML pipelines: increased inference costs arising from the need to generate 3D structures, limited training data, and robustness to noise in the generated structures. Conclusions: STAG-LLM shows tremendous potential for structure-based TCR-pHLA binding prediction methods, offering a foundation for further advancements in using modeled 3D structures to solve problems in immunology and proteomics. We anticipate that the usefulness of STAG-LLM and similar tools will increase in coming years as both protein structure prediction models and large language models continue to advance. - A. Conev, J. Chen, and L. E. Kavraki, “DINC-Ensemble: A Web Server for Docking Large Ligands Incrementally to an Ensemble of
Receptor Conformations,” Journal of Molecular Biology, p. 169163, Aug. 2025.
BibTeX
@article{conev2025-dinc-ensemble, title = {DINC-Ensemble: A Web Server for Docking Large Ligands Incrementally to an Ensemble of Receptor Conformations}, journal = {Journal of Molecular Biology}, pages = {169163}, year = {2025}, month = aug, issn = {0022-2836}, doi = {10.1016/j.jmb.2025.169163}, url = {https://www.sciencedirect.com/science/article/pii/S0022283625002293}, author = {Conev, Anja and Chen, Jing and Kavraki, Lydia E.}, keywords = {protein-ligand docking, ensemble docking, conformational selection, molecular docking web server}, abstract = {Protein-ligand docking aids structure-based drug discovery by computationally modelling protein-ligand interactions. DINC (Docking INCrementally) is one approach to molecular docking that improved the docking of large ligands using a parallelized incremental meta-docking. Traditional docking tools, including DINC, explore the flexibility of the ligand in a single receptor binding pocket assuming limited flexibility of the receptor backbone. This simplifying assumption narrows down the docking search space but hinders successful docking for flexible receptors. DINC-Ensemble implicitly considers receptor backbone flexibility by running DINC docking in parallel on different receptor conformations. Inputs to DINC-Ensemble include (1) a ligand and (2) a list of different receptor conformations. For each ligand-receptor pair DINC-Ensemble performs incremental meta-docking in parallel. As a result, multiple ligand poses are generated in the binding pockets of different receptor conformations. These poses are then ranked, and the lowest scoring pose is selected. Two main outputs provided by a successful run of DINC-Ensemble are (1) the best scoring ligand poses and (2) a ranked list of selected receptor conformations. The best scoring ligand pose can be used to understand the interactions between the receptor and the ligand that influence the binding. The ranked list of receptor conformations shows the best receptor conformation fit for a given ligand and can provide insight into ligand-induced conformational selection. We provide DINC-Ensemble as a Python package and a free web server athttps://dinc-ensemble.kavrakilab.rice.edu/.} }DetailsAbstract
Protein-ligand docking aids structure-based drug discovery by computationally modelling protein-ligand interactions. DINC (Docking INCrementally) is one approach to molecular docking that improved the docking of large ligands using a parallelized incremental meta-docking. Traditional docking tools, including DINC, explore the flexibility of the ligand in a single receptor binding pocket assuming limited flexibility of the receptor backbone. This simplifying assumption narrows down the docking search space but hinders successful docking for flexible receptors. DINC-Ensemble implicitly considers receptor backbone flexibility by running DINC docking in parallel on different receptor conformations. Inputs to DINC-Ensemble include (1) a ligand and (2) a list of different receptor conformations. For each ligand-receptor pair DINC-Ensemble performs incremental meta-docking in parallel. As a result, multiple ligand poses are generated in the binding pockets of different receptor conformations. These poses are then ranked, and the lowest scoring pose is selected. Two main outputs provided by a successful run of DINC-Ensemble are (1) the best scoring ligand poses and (2) a ranked list of selected receptor conformations. The best scoring ligand pose can be used to understand the interactions between the receptor and the ligand that influence the binding. The ranked list of receptor conformations shows the best receptor conformation fit for a given ligand and can provide insight into ligand-induced conformational selection. We provide DINC-Ensemble as a Python package and a free web server athttps://dinc-ensemble.kavrakilab.rice.edu/. - J. K. Slone, A. Conev, M. M. Rigo, A. Reuben, and L. E. Kavraki, “TCR-pMHC Binding Specificity Prediction from Structure Using Graph Neural Networks,” IEEE Transactions on Computational Biology and Bioinformatics, Jan. 2025.
BibTeX
@article{slone2025-tcr-binding, author = {Slone, Jared K. and Conev, Anja and Rigo, Mauricio M. and Reuben, Alexandre and Kavraki, Lydia E.}, title = {TCR-pMHC Binding Specificity Prediction from Structure Using Graph Neural Networks}, doi = {10.1109/TCBBIO.2024.3504235}, url = {http://ieeexplore.ieee.org/document/10819951}, journal = {IEEE Transactions on Computational Biology and Bioinformatics}, year = {2025}, month = jan, abstract = {The mapping of T-cell-receptors (TCRs) to their cognate peptides is crucial to improving cancer immunotherapy. Numerous computational methods and machine learning tools have been proposed to aid in the task. Yet, accurately constructing this map computationally remains a difficult problem. Most prior work has sought to predict TCR-peptide-MHC (TCR-pMHC) binding specificity by analyzing the amino acid sequences of the TCRs and peptides. However, recent advancements in crystallography, cryo-EM, and in silico protein modeling have provided researchers with the necessary data to analyze the 3D structures of TCRs, peptides, and MHCs. Current research suggests that information contained in the 3D structure of the TCRs and pMHCs can explain instances of TCR specificity that are not explained by sequence alone. As protein structure data continues to become more accurate and easier to obtain, structure-based methodologies for predicting TCR-pMHC binding will become increasingly important. We present STAG, a novel graph-based machine learning architecture for predicting TCR-pMHC binding specificity using 3D structure data. We show that STAG achieves comparable or better performance than existing methods while utilizing only spatial and physicochemical features from modeled protein structures.} }DetailsAbstract
The mapping of T-cell-receptors (TCRs) to their cognate peptides is crucial to improving cancer immunotherapy. Numerous computational methods and machine learning tools have been proposed to aid in the task. Yet, accurately constructing this map computationally remains a difficult problem. Most prior work has sought to predict TCR-peptide-MHC (TCR-pMHC) binding specificity by analyzing the amino acid sequences of the TCRs and peptides. However, recent advancements in crystallography, cryo-EM, and in silico protein modeling have provided researchers with the necessary data to analyze the 3D structures of TCRs, peptides, and MHCs. Current research suggests that information contained in the 3D structure of the TCRs and pMHCs can explain instances of TCR specificity that are not explained by sequence alone. As protein structure data continues to become more accurate and easier to obtain, structure-based methodologies for predicting TCR-pMHC binding will become increasingly important. We present STAG, a novel graph-based machine learning architecture for predicting TCR-pMHC binding specificity using 3D structure data. We show that STAG achieves comparable or better performance than existing methods while utilizing only spatial and physicochemical features from modeled protein structures. - R. Fasoulis, G. Paliouras, and L. E. Kavraki, “RankMHC: Learning to Rank Class-I Peptide-MHC Structural Models,” Journal of Chemical Information and Modeling, vol. 64, no. 23, pp. 8729–8742, Nov. 2024.
BibTeX
@article{fasoulis2024-rankmhc, author = {Fasoulis, Romanos and Paliouras, Georgios and Kavraki, Lydia E.}, title = {RankMHC: Learning to Rank Class-I Peptide-MHC Structural Models}, journal = {Journal of Chemical Information and Modeling}, volume = {64}, number = {23}, pages = {8729-8742}, year = {2024}, month = nov, doi = {10.1021/acs.jcim.4c01278}, abstract = {The binding of peptides to class-I Major Histocompability Complex (MHC) receptors and their subsequent recognition downstream by T-cell receptors are crucial processes for most multicellular organisms to be able to fight various diseases. Thus, the identification of peptide antigens that can elicit an immune response is of immense importance for developing successful therapies for bacterial and viral infections, even cancer. Recently, studies have demonstrated the importance of peptide-MHC (pMHC) structural analysis, with pMHC structural modeling methods gradually becoming more popular in peptide antigen identification workflows. Most of the pMHC structural modeling tools provide an ensemble of candidate peptide poses in the MHC-I cleft, each associated with a score stemming from a scoring function, with the top scoring pose assumed to be the most representative of the ensemble. However, identifying the binding mode, that is, the peptide pose from the ensemble that is closer to an unavailable native structure, is not trivial. Oftentimes, the peptide poses characterized as best by a protein–ligand scoring function are not the ones that are the most representative of the actual structure. In this work, we frame the peptide binding pose identification problem as a Learning-to-Rank (LTR) problem. We present RankMHC, an LTR-based pMHC binding mode identification predictor, which is specifically trained to predict the most accurate ranking of an ensemble of pMHC conformations. RankMHC outperforms classical peptide-ligand scoring functions, as well as previous Machine Learning (ML)-based binding pose predictors. We further demonstrate that RankMHC can be used with many pMHC structural modeling tools that use different structural modeling protocols.} }DetailsAbstract
The binding of peptides to class-I Major Histocompability Complex (MHC) receptors and their subsequent recognition downstream by T-cell receptors are crucial processes for most multicellular organisms to be able to fight various diseases. Thus, the identification of peptide antigens that can elicit an immune response is of immense importance for developing successful therapies for bacterial and viral infections, even cancer. Recently, studies have demonstrated the importance of peptide-MHC (pMHC) structural analysis, with pMHC structural modeling methods gradually becoming more popular in peptide antigen identification workflows. Most of the pMHC structural modeling tools provide an ensemble of candidate peptide poses in the MHC-I cleft, each associated with a score stemming from a scoring function, with the top scoring pose assumed to be the most representative of the ensemble. However, identifying the binding mode, that is, the peptide pose from the ensemble that is closer to an unavailable native structure, is not trivial. Oftentimes, the peptide poses characterized as best by a protein–ligand scoring function are not the ones that are the most representative of the actual structure. In this work, we frame the peptide binding pose identification problem as a Learning-to-Rank (LTR) problem. We present RankMHC, an LTR-based pMHC binding mode identification predictor, which is specifically trained to predict the most accurate ranking of an ensemble of pMHC conformations. RankMHC outperforms classical peptide-ligand scoring functions, as well as previous Machine Learning (ML)-based binding pose predictors. We further demonstrate that RankMHC can be used with many pMHC structural modeling tools that use different structural modeling protocols. - R. Fasoulis, M. M. Rigo, G. Lizée, D. A. Antunes, and L. E. Kavraki, “APE-Gen2.0: Expanding Rapid Class I Peptide-Major Histocompatibility Complex
Modeling to Post-Translational Modifications and Noncanonical Peptide Geometries,” Journal of Chemical Information and Modeling, vol. 64, no. 5, pp. 1730–1750, Mar. 2024.
BibTeX
@article{fasoulis2024-apegen2, title = {{APE-Gen2.0}: Expanding Rapid Class {I} Peptide-Major Histocompatibility Complex Modeling to Post-Translational Modifications and Noncanonical Peptide Geometries}, author = {Fasoulis, Romanos and Rigo, Mauricio M. and Liz{\'e}e, Gregory and Antunes, Dinler A. and Kavraki, Lydia E.}, journal = {Journal of Chemical Information and Modeling}, year = {2024}, doi = {10.1021/acs.jcim.3c01667}, url = {https://pubs.acs.org/doi/10.1021/acs.jcim.3c01667}, month = mar, pages = {1730-1750}, volume = {64}, issue = {5}, keywords = {Animals, *Peptides/chemistry, Major Histocompatibility Complex, Receptors, Antigen, T-Cell/genetics/metabolism, Protein Processing, Post-Translational, *Hominidae/metabolism, Protein Binding}, abstract = {The recognition of peptides bound to class I major histocompatibility complex (MHC-I) receptors by T-cell receptors (TCRs) is a determinant of triggering the adaptive immune response. While the exact molecular features that drive the TCR recognition are still unknown, studies have suggested that the geometry of the joint peptide-MHC (pMHC) structure plays an important role. As such, there is a definite need for methods and tools that accurately predict the structure of the peptide bound to the MHC-I receptor. In the past few years, many pMHC structural modeling tools have emerged that provide high-quality modeled structures in the general case. However, there are numerous instances of non-canonical cases in the immunopeptidome that the majority of pMHC modeling tools do not attend to, most notably, peptides that exhibit non-standard amino acids and post-translational modifications (PTMs) or peptides that assume non-canonical geometries in the MHC binding cleft. Such chemical and structural properties have been shown to be present in neoantigens; therefore, accurate structural modeling of these instances can be vital for cancer immunotherapy. To this end, we have developed APE-Gen2.0, a tool that improves upon its predecessor and other pMHC modeling tools, both in terms of modeling accuracy and the available modeling range of non-canonical peptide cases. Some of the improvements include (i) the ability to model peptides that have different types of PTMs such as phosphorylation, nitration, and citrullination; (ii) a new and improved anchor identification routine in order to identify and model peptides that exhibit a non-canonical anchor conformation; and (iii) a web server that provides a platform for easy and accessible pMHC modeling. We further show that structures predicted by APE-Gen2.0 can be used to assess the effects that PTMs have in binding affinity in a more accurate manner than just using solely the sequence of the peptide. APE-Gen2.0 is freely available at https://apegen.kavrakilab.org.} }DetailsAbstract
The recognition of peptides bound to class I major histocompatibility complex (MHC-I) receptors by T-cell receptors (TCRs) is a determinant of triggering the adaptive immune response. While the exact molecular features that drive the TCR recognition are still unknown, studies have suggested that the geometry of the joint peptide-MHC (pMHC) structure plays an important role. As such, there is a definite need for methods and tools that accurately predict the structure of the peptide bound to the MHC-I receptor. In the past few years, many pMHC structural modeling tools have emerged that provide high-quality modeled structures in the general case. However, there are numerous instances of non-canonical cases in the immunopeptidome that the majority of pMHC modeling tools do not attend to, most notably, peptides that exhibit non-standard amino acids and post-translational modifications (PTMs) or peptides that assume non-canonical geometries in the MHC binding cleft. Such chemical and structural properties have been shown to be present in neoantigens; therefore, accurate structural modeling of these instances can be vital for cancer immunotherapy. To this end, we have developed APE-Gen2.0, a tool that improves upon its predecessor and other pMHC modeling tools, both in terms of modeling accuracy and the available modeling range of non-canonical peptide cases. Some of the improvements include (i) the ability to model peptides that have different types of PTMs such as phosphorylation, nitration, and citrullination; (ii) a new and improved anchor identification routine in order to identify and model peptides that exhibit a non-canonical anchor conformation; and (iii) a web server that provides a platform for easy and accessible pMHC modeling. We further show that structures predicted by APE-Gen2.0 can be used to assess the effects that PTMs have in binding affinity in a more accurate manner than just using solely the sequence of the peptide. APE-Gen2.0 is freely available at https://apegen.kavrakilab.org. - R. Fasoulis, M. M. Rigo, D. A. Antunes, G. Paliouras, and L. E. Kavraki, “Transfer learning improves pMHC kinetic stability and immunogenicity predictions,” ImmunoInformatics, vol. 13, p. 100030, Mar. 2024.
BibTeX
@article{fasoulis2024-transfer, title = {Transfer learning improves pMHC kinetic stability and immunogenicity predictions}, journal = {ImmunoInformatics}, volume = {13}, pages = {100030}, year = {2024}, month = mar, issn = {2667-1190}, doi = {10.1016/j.immuno.2023.100030}, url = {https://www.sciencedirect.com/science/article/pii/S2667119023000101}, author = {Fasoulis, Romanos and Rigo, Mauricio Menegatti and Antunes, Dinler Amaral and Paliouras, Georgios and Kavraki, Lydia E.}, keywords = {Transfer learning, Peptide-MHC, Machine learning, Peptide kinetic stability, Peptide immunogenicity}, abstract = {The cellular immune response comprises several processes, with the most notable ones being the binding of the peptide to the Major Histocompability Complex (MHC), the peptide-MHC (pMHC) presentation to the surface of the cell, and the recognition of the pMHC by the T-Cell Receptor. Identifying the most potent peptide targets for MHC binding, presentation and T-cell recognition is vital for developing peptide-based vaccines and T-cell-based immunotherapies. Data-driven tools that predict each of these steps have been developed, and the availability of mass spectrometry (MS) datasets has facilitated the development of accurate Machine Learning (ML) methods for class-I pMHC binding prediction. However, the accuracy of ML-based tools for pMHC kinetic stability prediction and peptide immunogenicity prediction is uncertain, as stability and immunogenicity datasets are not abundant. Here, we use transfer learning techniques to improve stability and immunogenicity predictions, by taking advantage of a large number of binding affinity and MS datasets. The resulting models, TLStab and TLImm, exhibit comparable or better performance than state-of-the-art approaches on different stability and immunogenicity test sets respectively. Our approach demonstrates the promise of learning from the task of peptide binding to improve predictions on downstream tasks. The source code of TLStab and TLImm is publicly available at https://github.com/KavrakiLab/TL-MHC.} }DetailsAbstract
The cellular immune response comprises several processes, with the most notable ones being the binding of the peptide to the Major Histocompability Complex (MHC), the peptide-MHC (pMHC) presentation to the surface of the cell, and the recognition of the pMHC by the T-Cell Receptor. Identifying the most potent peptide targets for MHC binding, presentation and T-cell recognition is vital for developing peptide-based vaccines and T-cell-based immunotherapies. Data-driven tools that predict each of these steps have been developed, and the availability of mass spectrometry (MS) datasets has facilitated the development of accurate Machine Learning (ML) methods for class-I pMHC binding prediction. However, the accuracy of ML-based tools for pMHC kinetic stability prediction and peptide immunogenicity prediction is uncertain, as stability and immunogenicity datasets are not abundant. Here, we use transfer learning techniques to improve stability and immunogenicity predictions, by taking advantage of a large number of binding affinity and MS datasets. The resulting models, TLStab and TLImm, exhibit comparable or better performance than state-of-the-art approaches on different stability and immunogenicity test sets respectively. Our approach demonstrates the promise of learning from the task of peptide binding to improve predictions on downstream tasks. The source code of TLStab and TLImm is publicly available at https://github.com/KavrakiLab/TL-MHC. - A. Conev, R. Fasoulis, S. Hall-Swan, R. Ferreira, and L. E. Kavraki, “HLAEquity: Examining biases in pan-allele peptide-HLA binding predictors,” iScience, vol. 27, no. 1, p. 108613, Jan. 2024.
BibTeX
@article{conev2024-hlaequity, author = {Conev, Anja and Fasoulis, Romanos and Hall-Swan, Sarah and Ferreira, Rodrigo and Kavraki, Lydia E}, title = {{HLAEquity: Examining biases in pan-allele peptide-HLA binding predictors}}, journal = {iScience}, year = {2024}, month = jan, volume = {27}, number = {1}, pages = {108613}, abstract = {Peptide-HLA (pHLA) binding prediction is essential in screening peptide candidates for personalized peptide vaccines. Machine Learning (ML) pHLA binding prediction tools are trained on vast amounts of data and are effective in screening peptide candidates. Most ML models report generalizing to HLA alleles unseen during training (“pan-allele” models). However, the use of datasets with imbalanced allele content raises concerns about biased model performance. First, we examine the data bias of two ML-based pan-allele pHLA binding predictors. We find that the pHLA datasets overrepresent alleles from geographic populations of high-income countries. Second, we show that the identified data bias is perpetuated within ML models, leading to algorithmic bias and subpar performance for alleles expressed in low-income geographic populations. We draw attention to the potential therapeutic consequences of this bias, and we challenge the use of the term “pan-allele” to describe models trained with currently available public datasets.}, issn = {2589-0042}, doi = {10.1016/j.isci.2023.108613}, url = {https://doi.org/10.1016/j.isci.2023.108613}, eprint = {https://www.sciencedirect.com/science/article/pii/S2589004223026901} }DetailsAbstract
Peptide-HLA (pHLA) binding prediction is essential in screening peptide candidates for personalized peptide vaccines. Machine Learning (ML) pHLA binding prediction tools are trained on vast amounts of data and are effective in screening peptide candidates. Most ML models report generalizing to HLA alleles unseen during training (“pan-allele” models). However, the use of datasets with imbalanced allele content raises concerns about biased model performance. First, we examine the data bias of two ML-based pan-allele pHLA binding predictors. We find that the pHLA datasets overrepresent alleles from geographic populations of high-income countries. Second, we show that the identified data bias is perpetuated within ML models, leading to algorithmic bias and subpar performance for alleles expressed in low-income geographic populations. We draw attention to the potential therapeutic consequences of this bias, and we challenge the use of the term “pan-allele” to describe models trained with currently available public datasets. - A. Conev, M. M. Rigo, D. Devaurs, A. F. Fonseca, H. Kalavadwala, M. V. de Freitas, C. Clementi, G. Zanatta, D. A. Antunes, and L. E. Kavraki, “EnGens: a computational framework for generation and analysis of representative
protein conformational ensembles,” Briefings in Bioinformatics, vol. 24, no. 4, p. bbad242, Jul. 2023.
BibTeX
@article{conev2023-engens, author = {Conev, Anja and Rigo, Mauricio Menegatti and Devaurs, Didier and Fonseca, André Faustino and Kalavadwala, Hussain and de Freitas, Martiela Vaz and Clementi, Cecilia and Zanatta, Geancarlo and Antunes, Dinler Amaral and Kavraki, Lydia E}, title = {{EnGens: a computational framework for generation and analysis of representative protein conformational ensembles}}, journal = {Briefings in Bioinformatics}, pages = {bbad242}, year = {2023}, month = jul, volume = {24}, issue = {4}, abstract = {{Proteins are dynamic macromolecules that perform vital functions in cells. A protein structure determines its function, but this structure is not static, as proteins change their conformation to achieve various functions. Understanding the conformational landscapes of proteins is essential to understand their mechanism of action. Sets of carefully chosen conformations can summarize such complex landscapes and provide better insights into protein function than single conformations. We refer to these sets as representative conformational ensembles. Recent advances in computational methods have led to an increase in the number of available structural datasets spanning conformational landscapes. However, extracting representative conformational ensembles from such datasets is not an easy task and many methods have been developed to tackle it. Our new approach, EnGens (short for ensemble generation), collects these methods into a unified framework for generating and analyzing representative protein conformational ensembles. In this work, we: (1) provide an overview of existing methods and tools for representative protein structural ensemble generation and analysis; (2) unify existing approaches in an open-source Python package, and a portable Docker image, providing interactive visualizations within a Jupyter Notebook pipeline; (3) test our pipeline on a few canonical examples from the literature. Representative ensembles produced by EnGens can be used for many downstream tasks such as protein–ligand ensemble docking, Markov state modeling of protein dynamics and analysis of the effect of single-point mutations.}}, issn = {1477-4054}, doi = {10.1093/bib/bbad242}, url = {https://doi.org/10.1093/bib/bbad242}, eprint = {https://academic.oup.com/bib/advance-article-pdf/doi/10.1093/bib/bbad242/50837168/bbad242.pdf} }DetailsAbstract
Proteins are dynamic macromolecules that perform vital functions in cells. A protein structure determines its function, but this structure is not static, as proteins change their conformation to achieve various functions. Understanding the conformational landscapes of proteins is essential to understand their mechanism of action. Sets of carefully chosen conformations can summarize such complex landscapes and provide better insights into protein function than single conformations. We refer to these sets as representative conformational ensembles. Recent advances in computational methods have led to an increase in the number of available structural datasets spanning conformational landscapes. However, extracting representative conformational ensembles from such datasets is not an easy task and many methods have been developed to tackle it. Our new approach, EnGens (short for ensemble generation), collects these methods into a unified framework for generating and analyzing representative protein conformational ensembles. In this work, we: (1) provide an overview of existing methods and tools for representative protein structural ensemble generation and analysis; (2) unify existing approaches in an open-source Python package, and a portable Docker image, providing interactive visualizations within a Jupyter Notebook pipeline; (3) test our pipeline on a few canonical examples from the literature. Representative ensembles produced by EnGens can be used for many downstream tasks such as protein–ligand ensemble docking, Markov state modeling of protein dynamics and analysis of the effect of single-point mutations. - E. E. Litsa, V. Chenthamarakshan, P. Das, and L. E. Kavraki, “An end-to-end deep learning framework for translating mass spectra to de-novo
molecules,” Communications Chemistry, vol. 6, no. 1, p. 132, Jun. 2023.
BibTeX
@article{litsa2023spec2mol, title = {An end-to-end deep learning framework for translating mass spectra to de-novo molecules}, author = {Litsa, Eleni E and Chenthamarakshan, Vijil and Das, Payel and Kavraki, Lydia E}, journal = {Communications Chemistry}, volume = {6}, number = {1}, pages = {132}, year = {2023}, month = jun, publisher = {Nature Publishing Group UK London}, doi = {10.1038/s42004-023-00932-3}, abstract = {Elucidating the structure of a chemical compound is a fundamental task in chemistry with applications in multiple domains including drug discovery, precision medicine, and biomarker discovery. The common practice for elucidating the structure of a compound is to obtain a mass spectrum and subsequently retrieve its structure from spectral databases. However, these methods fail for novel molecules that are not present in the reference database. We propose Spec2Mol, a deep learning architecture for molecular structure recommendation given mass spectra alone. Spec2Mol is inspired by the Speech2Text deep learning architectures for translating audio signals into text. Our approach is based on an encoder-decoder architecture. The encoder learns the spectra embeddings, while the decoder, pre-trained on a massive dataset of chemical structures for translating between different molecular representations, reconstructs SMILES sequences of the recommended chemical structures. We have evaluated Spec2Mol by assessing the molecular similarity between the recommended structures and the original structure. Our analysis showed that Spec2Mol is able to identify the presence of key molecular substructures from its mass spectrum, and shows on par performance, when compared to existing fragmentation tree methods particularly when test structure information is not available during training or present in the reference database.} }DetailsAbstract
Elucidating the structure of a chemical compound is a fundamental task in chemistry with applications in multiple domains including drug discovery, precision medicine, and biomarker discovery. The common practice for elucidating the structure of a compound is to obtain a mass spectrum and subsequently retrieve its structure from spectral databases. However, these methods fail for novel molecules that are not present in the reference database. We propose Spec2Mol, a deep learning architecture for molecular structure recommendation given mass spectra alone. Spec2Mol is inspired by the Speech2Text deep learning architectures for translating audio signals into text. Our approach is based on an encoder-decoder architecture. The encoder learns the spectra embeddings, while the decoder, pre-trained on a massive dataset of chemical structures for translating between different molecular representations, reconstructs SMILES sequences of the recommended chemical structures. We have evaluated Spec2Mol by assessing the molecular similarity between the recommended structures and the original structure. Our analysis showed that Spec2Mol is able to identify the presence of key molecular substructures from its mass spectrum, and shows on par performance, when compared to existing fragmentation tree methods particularly when test structure information is not available during training or present in the reference database. - S. Hall-Swan, J. Slone, M. M. Rigo, D. A. Antunes, G. Lizée, and L. E. Kavraki, “PepSim: T-cell cross-reactivity prediction via comparison of peptide sequence and
peptide-HLA structure,” Frontiers in Immunology, vol. 14, Apr. 2023.
BibTeX
@article{hall-swan2023pepsim, author = {Hall-Swan, Sarah and Slone, Jared and Rigo, Mauricio M. and Antunes, Dinler A. and Lizée, Gregory and Kavraki, Lydia E.}, title = {PepSim: T-cell cross-reactivity prediction via comparison of peptide sequence and peptide-HLA structure}, journal = {Frontiers in Immunology}, volume = {14}, year = {2023}, month = apr, url = {https://www.frontiersin.org/articles/10.3389/fimmu.2023.1108303}, doi = {10.3389/fimmu.2023.1108303}, issn = {1664-3224}, abstract = {Introduction: Peptide-HLA class I (pHLA) complexes on the surface of tumor cells can be targeted by cytotoxic T-cells to eliminate tumors, and this is one of the bases for T-cell-based immunotherapies. However, there exist cases where therapeutic T-cells directed towards tumor pHLA complexes may also recognize pHLAs from healthy normal cells. The process where the same T-cell clone recognizes more than one pHLA is referred to as T-cell cross-reactivity and this process is driven mainly by features that make pHLAs similar to each other. T-cell cross-reactivity prediction is critical for designing T-cell-based cancer immunotherapies that are both effective and safe. Methods: Here we present PepSim, a novel score to predict T-cell cross-reactivity based on the structural and biochemical similarity of pHLAs. Results and discussion: We show our method can accurately separate cross-reactive from non-crossreactive pHLAs in a diverse set of datasets including cancer, viral, and self-peptides. PepSim can be generalized to work on any dataset of class I peptide-HLAs and is freely available as a web server at pepsim.kavrakilab.org.} }DetailsAbstract
Introduction: Peptide-HLA class I (pHLA) complexes on the surface of tumor cells can be targeted by cytotoxic T-cells to eliminate tumors, and this is one of the bases for T-cell-based immunotherapies. However, there exist cases where therapeutic T-cells directed towards tumor pHLA complexes may also recognize pHLAs from healthy normal cells. The process where the same T-cell clone recognizes more than one pHLA is referred to as T-cell cross-reactivity and this process is driven mainly by features that make pHLAs similar to each other. T-cell cross-reactivity prediction is critical for designing T-cell-based cancer immunotherapies that are both effective and safe. Methods: Here we present PepSim, a novel score to predict T-cell cross-reactivity based on the structural and biochemical similarity of pHLAs. Results and discussion: We show our method can accurately separate cross-reactive from non-crossreactive pHLAs in a diverse set of datasets including cancer, viral, and self-peptides. PepSim can be generalized to work on any dataset of class I peptide-HLAs and is freely available as a web server at pepsim.kavrakilab.org. - K. R. Jackson, D. A. Antunes, A. H. Talukder, A. R. Maleki, K. Amagai, A. Salmon, A. S. Katailiha, Y. Chiu, R. Fasoulis, M. M. Rigo, J. R. Abella, B. D. Melendez, F. Li, Y. Sun, H. M. Sonnemann, V. Belousov, F. Frenkel, S. Justesen, A. Makaju, Y. Liu, D. Horn, D. Lopez-Ferrer, A. F. Huhmer, P. Hwu, J. Roszik, D. Hawke, L. E. Kavraki, and G. Lizée, “Charge-based interactions through peptide position 4 drive diversity of antigen
presentation by human leukocyte antigen class I molecules,” PNAS Nexus, vol. 1, no. 3, Aug. 2022.
BibTeX
@article{jackson2022-charge-interactions, author = {Jackson, Kyle R and Antunes, Dinler A and Talukder, Amjad H and Maleki, Ariana R and Amagai, Kano and Salmon, Avery and Katailiha, Arjun S and Chiu, Yulun and Fasoulis, Romanos and Rigo, Maurício Menegatti and Abella, Jayvee R and Melendez, Brenda D and Li, Fenge and Sun, Yimo and Sonnemann, Heather M and Belousov, Vladislav and Frenkel, Felix and Justesen, Sune and Makaju, Aman and Liu, Yang and Horn, David and Lopez-Ferrer, Daniel and Huhmer, Andreas F and Hwu, Patrick and Roszik, Jason and Hawke, David and Kavraki, Lydia E and Lizée, Gregory}, title = {{Charge-based interactions through peptide position 4 drive diversity of antigen presentation by human leukocyte antigen class I molecules}}, journal = {PNAS Nexus}, volume = {1}, number = {3}, year = {2022}, month = aug, abstract = {Human leukocyte antigen class I (HLA-I) molecules bind and present peptides at the cell surface to facilitate the induction of appropriate CD8+ T cell-mediated immune responses to pathogen- and self-derived proteins. The HLA-I peptide-binding cleft contains dominant anchor sites in the B and F pockets that interact primarily with amino acids at peptide position 2 and the C-terminus, respectively. Nonpocket peptide–HLA interactions also contribute to peptide binding and stability, but these secondary interactions are thought to be unique to individual HLA allotypes or to specific peptide antigens. Here, we show that two positively charged residues located near the top of peptide-binding cleft facilitate interactions with negatively charged residues at position 4 of presented peptides, which occur at elevated frequencies across most HLA-I allotypes. Loss of these interactions was shown to impair HLA-I/peptide binding and complex stability, as demonstrated by both in vitro and in silico experiments. Furthermore, mutation of these Arginine-65 (R65) and/or Lysine-66 (K66) residues in HLA-A*02:01 and A*24:02 significantly reduced HLA-I cell surface expression while also reducing the diversity of the presented peptide repertoire by up to 5-fold. The impact of the R65 mutation demonstrates that nonpocket HLA-I/peptide interactions can constitute anchor motifs that exert an unexpectedly broad influence on HLA-I-mediated antigen presentation. These findings provide fundamental insights into peptide antigen binding that could broadly inform epitope discovery in the context of viral vaccine development and cancer immunotherapy.}, issn = {2752-6542}, doi = {10.1093/pnasnexus/pgac124}, url = {https://doi.org/10.1093/pnasnexus/pgac124} }DetailsAbstract
Human leukocyte antigen class I (HLA-I) molecules bind and present peptides at the cell surface to facilitate the induction of appropriate CD8+ T cell-mediated immune responses to pathogen- and self-derived proteins. The HLA-I peptide-binding cleft contains dominant anchor sites in the B and F pockets that interact primarily with amino acids at peptide position 2 and the C-terminus, respectively. Nonpocket peptide–HLA interactions also contribute to peptide binding and stability, but these secondary interactions are thought to be unique to individual HLA allotypes or to specific peptide antigens. Here, we show that two positively charged residues located near the top of peptide-binding cleft facilitate interactions with negatively charged residues at position 4 of presented peptides, which occur at elevated frequencies across most HLA-I allotypes. Loss of these interactions was shown to impair HLA-I/peptide binding and complex stability, as demonstrated by both in vitro and in silico experiments. Furthermore, mutation of these Arginine-65 (R65) and/or Lysine-66 (K66) residues in HLA-A*02:01 and A*24:02 significantly reduced HLA-I cell surface expression while also reducing the diversity of the presented peptide repertoire by up to 5-fold. The impact of the R65 mutation demonstrates that nonpocket HLA-I/peptide interactions can constitute anchor motifs that exert an unexpectedly broad influence on HLA-I-mediated antigen presentation. These findings provide fundamental insights into peptide antigen binding that could broadly inform epitope discovery in the context of viral vaccine development and cancer immunotherapy. - M. M. Rigo, R. Fasoulis, A. Conev, S. Hall-Swan, D. Amaral Antunes, and L. Kavraki, “SARS-Arena: Sequence and Structure-Guided Selection of Conserved Peptides from
SARS-related Coronaviruses for Novel Vaccine Development,” Frontiers in Immunology, vol. 13, Jul. 2022.
BibTeX
@article{rigo2022-sars-arena, title = {SARS-Arena: Sequence and Structure-Guided Selection of Conserved Peptides from SARS-related Coronaviruses for Novel Vaccine Development}, author = {Rigo, Mauricio Menegatti and Fasoulis, Romanos and Conev, Anja and Hall-Swan, Sarah and Amaral Antunes, Dinler and Kavraki, Lydia}, journal = {Frontiers in Immunology}, month = jul, year = {2022}, volume = {13}, doi = {10.3389/fimmu.2022.931155}, abstract = {The pandemic caused by the SARS-CoV-2 virus, the agent responsible for the COVID-19 disease, has affected millions of people worldwide. There is constant search for new therapies to either prevent or mitigate the disease. Fortunately, we have observed the successful development of multiple vaccines. Most of them are focused on one viral envelope protein, the spike protein. However, such focused approaches may contribute for the rise of new variants, fueled by the constant selection pressure on envelope proteins, and the widespread dispersion of coronaviruses in nature. Therefore, it is important to examine other proteins, preferentially those that are less susceptible to selection pressure, such as the nucleocapsid (N) protein. Even though the N protein is less accessible to humoral response, peptides from its conserved regions can be presented by class I Human Leukocyte Antigen (HLA) molecules, eliciting an immune response mediated by T-cells. Given the increased number of protein sequences deposited in biological databases daily and the N protein conservation among viral strains, computational methods can be leveraged to discover potential new targets for SARS-CoV-2 and SARS-CoV-related viruses. Here we developed SARS-Arena, a user-friendly computational pipeline that can be used by practitioners of different levels of expertise for novel vaccine development. SARS-Arena combines sequence-based methods and structure-based analyses to (i) perform multiple sequence alignment (MSA) of SARS-CoV-related N protein sequences, (ii) recover candidate peptides of different lengths from conserved protein regions, and (iii) model the 3D structure of the conserved peptides in the context of different HLAs. We present two main Jupyter Notebook workflows that can help in the identification of new T-cell targets against SARS-CoV viruses. In fact, in a cross-reactive case study, our workflows identified a conserved N protein peptide (SPRWYFYYL) recognized by CD8+ T-cells in the context of HLA-B7+. SARS-Arena is available at https://github.com/KavrakiLab/SARS-Arena.}, publisher = {Frontiers Media SA}, url = {https://doi.org/10.3389/fimmu.2022.931155} }DetailsAbstract
The pandemic caused by the SARS-CoV-2 virus, the agent responsible for the COVID-19 disease, has affected millions of people worldwide. There is constant search for new therapies to either prevent or mitigate the disease. Fortunately, we have observed the successful development of multiple vaccines. Most of them are focused on one viral envelope protein, the spike protein. However, such focused approaches may contribute for the rise of new variants, fueled by the constant selection pressure on envelope proteins, and the widespread dispersion of coronaviruses in nature. Therefore, it is important to examine other proteins, preferentially those that are less susceptible to selection pressure, such as the nucleocapsid (N) protein. Even though the N protein is less accessible to humoral response, peptides from its conserved regions can be presented by class I Human Leukocyte Antigen (HLA) molecules, eliciting an immune response mediated by T-cells. Given the increased number of protein sequences deposited in biological databases daily and the N protein conservation among viral strains, computational methods can be leveraged to discover potential new targets for SARS-CoV-2 and SARS-CoV-related viruses. Here we developed SARS-Arena, a user-friendly computational pipeline that can be used by practitioners of different levels of expertise for novel vaccine development. SARS-Arena combines sequence-based methods and structure-based analyses to (i) perform multiple sequence alignment (MSA) of SARS-CoV-related N protein sequences, (ii) recover candidate peptides of different lengths from conserved protein regions, and (iii) model the 3D structure of the conserved peptides in the context of different HLAs. We present two main Jupyter Notebook workflows that can help in the identification of new T-cell targets against SARS-CoV viruses. In fact, in a cross-reactive case study, our workflows identified a conserved N protein peptide (SPRWYFYYL) recognized by CD8+ T-cells in the context of HLA-B7+. SARS-Arena is available at https://github.com/KavrakiLab/SARS-Arena. - A. Conev, D. Devaurs, M. M. Rigo, D. A. Antunes, and L. E. Kavraki, “3pHLA-score improves structure-based peptide-HLA binding affinity prediction,” Scientific Reports, vol. 12, no. 1, Jun. 2022.
BibTeX
@article{conev2022-3phla-score, title = {3pHLA-score improves structure-based peptide-{HLA} binding affinity prediction}, author = {Conev, Anja and Devaurs, Didier and Rigo, Mauricio M. and Antunes, Dinler A. and Kavraki, Lydia E.}, journal = {Scientific Reports}, month = jun, year = {2022}, volume = {12}, number = {1}, doi = {10.1038/s41598-022-14526-x}, abstract = {Binding of peptides to Human Leukocyte Antigen (HLA) receptors is a prerequisite for triggering immune response. Estimating peptide-HLA (pHLA) binding is crucial for peptide vaccine target identification and epitope discovery pipelines. Computational methods for binding affinity prediction can accelerate these pipelines. Currently, most of those computational methods rely exclusively on sequence-based data, which leads to inherent limitations. Recent studies have shown that structure-based data can address some of these limitations. In this work we propose a novel machine learning (ML) structure-based protocol to predict binding affinity of peptides to HLA receptors. For that, we engineer the input features for ML models by decoupling energy contributions at different residue positions in peptides, which leads to our novel per-peptide-position protocol. Using Rosetta’s ref2015 scoring function as a baseline we use this protocol to develop 3pHLA-score. Our per-peptide-position protocol outperforms the standard training protocol and leads to an increase from 0.82 to 0.99 of the area under the precision-recall curve. 3pHLA-score outperforms widely used scoring functions (AutoDock4, Vina, Dope, Vinardo, FoldX, GradDock) in a structural virtual screening task. Overall, this work brings structure-based methods one step closer to epitope discovery pipelines and could help advance the development of cancer and viral vaccines.}, keyword = {proteins and drugs}, publisher = {Springer Science and Business Media {LLC}}, url = {https://doi.org/10.1038/s41598-022-14526-x} }DetailsAbstract
Binding of peptides to Human Leukocyte Antigen (HLA) receptors is a prerequisite for triggering immune response. Estimating peptide-HLA (pHLA) binding is crucial for peptide vaccine target identification and epitope discovery pipelines. Computational methods for binding affinity prediction can accelerate these pipelines. Currently, most of those computational methods rely exclusively on sequence-based data, which leads to inherent limitations. Recent studies have shown that structure-based data can address some of these limitations. In this work we propose a novel machine learning (ML) structure-based protocol to predict binding affinity of peptides to HLA receptors. For that, we engineer the input features for ML models by decoupling energy contributions at different residue positions in peptides, which leads to our novel per-peptide-position protocol. Using Rosetta’s ref2015 scoring function as a baseline we use this protocol to develop 3pHLA-score. Our per-peptide-position protocol outperforms the standard training protocol and leads to an increase from 0.82 to 0.99 of the area under the precision-recall curve. 3pHLA-score outperforms widely used scoring functions (AutoDock4, Vina, Dope, Vinardo, FoldX, GradDock) in a structural virtual screening task. Overall, this work brings structure-based methods one step closer to epitope discovery pipelines and could help advance the development of cancer and viral vaccines. - R. F. Tarabini, M. M. Rigo, A. Faustino Fonseca, F. Rubin, R. Bellé, L. E. Kavraki, T. C. Ferreto, D. Amaral Antunes, and A. P. D. de Souza, “Large-Scale Structure-Based Screening of Potential T Cell Cross-Reactivities Involving
Peptide-Targets From BCG Vaccine and SARS-CoV-2,” Frontiers in Immunology, vol. 12, Jan. 2022.
BibTeX
@article{tarabini2022-large-scale, title = {Large-Scale Structure-Based Screening of Potential T Cell Cross-Reactivities Involving Peptide-Targets From BCG Vaccine and SARS-CoV-2}, author = {Tarabini, Renata Fioravanti and Rigo, Mauricio Menegatti and Faustino Fonseca, André and Rubin, Felipe and Bellé, Rafael and Kavraki, Lydia E and Ferreto, Tiago Coelho and Amaral Antunes, Dinler and de Souza, Ana Paula Duarte}, journal = {Frontiers in Immunology}, month = jan, year = {2022}, volume = {12}, doi = {10.3389/fimmu.2021.812176}, abstract = {Although not being the first viral pandemic to affect humankind, we are now for the first time faced with a pandemic caused by a coronavirus. The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has been responsible for the COVID-19 pandemic, which caused more than 4.5 million deaths worldwide. Despite unprecedented efforts, with vaccines being developed in a record time, SARS-CoV-2 continues to spread worldwide with new variants arising in different countries. Such persistent spread is in part enabled by public resistance to vaccination in some countries, and limited access to vaccines in other countries. The limited vaccination coverage, the continued risk for resistant variants, and the existence of natural reservoirs for coronaviruses, highlight the importance of developing additional therapeutic strategies against SARS-CoV-2 and other coronaviruses. At the beginning of the pandemic it was suggested that countries with Bacillus Calmette-Guérin (BCG) vaccination programs could be associated with a reduced number and/or severity of COVID-19 cases. Preliminary studies have provided evidence for this relationship and further investigation is being conducted in ongoing clinical trials. The protection against SARS-CoV-2 induced by BCG vaccination may be mediated by cross-reactive T cell lymphocytes, which recognize peptides displayed by class I Human Leukocyte Antigens (HLA-I) on the surface of infected cells. In order to identify potential targets of T cell cross-reactivity, we implemented an in silico strategy combining sequence-based and structure-based methods to screen over 13,5 million possible cross-reactive peptide pairs from BCG and SARS-CoV-2. Our study produced (i) a list of immunogenic BCG-derived peptides that may prime T cell cross-reactivity against SARS-CoV-2, (ii) a large dataset of modeled peptide-HLA structures for the screened targets, and (iii) new computational methods for structure-based screenings that can be used by others in future studies. Our study expands the list of BCG peptides potentially involved in T cell cross-reactivity with SARS-CoV-2-derived peptides, and identifies multiple high-density "neighborhoods" of cross-reactive peptides which could be driving heterologous immunity induced by BCG vaccination, therefore providing insights for future vaccine development efforts.}, issn = {1664-3224}, publisher = {Frontiers Media SA}, url = {http://dx.doi.org/10.3389/fimmu.2021.812176} }DetailsAbstract
Although not being the first viral pandemic to affect humankind, we are now for the first time faced with a pandemic caused by a coronavirus. The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has been responsible for the COVID-19 pandemic, which caused more than 4.5 million deaths worldwide. Despite unprecedented efforts, with vaccines being developed in a record time, SARS-CoV-2 continues to spread worldwide with new variants arising in different countries. Such persistent spread is in part enabled by public resistance to vaccination in some countries, and limited access to vaccines in other countries. The limited vaccination coverage, the continued risk for resistant variants, and the existence of natural reservoirs for coronaviruses, highlight the importance of developing additional therapeutic strategies against SARS-CoV-2 and other coronaviruses. At the beginning of the pandemic it was suggested that countries with Bacillus Calmette-Guérin (BCG) vaccination programs could be associated with a reduced number and/or severity of COVID-19 cases. Preliminary studies have provided evidence for this relationship and further investigation is being conducted in ongoing clinical trials. The protection against SARS-CoV-2 induced by BCG vaccination may be mediated by cross-reactive T cell lymphocytes, which recognize peptides displayed by class I Human Leukocyte Antigens (HLA-I) on the surface of infected cells. In order to identify potential targets of T cell cross-reactivity, we implemented an in silico strategy combining sequence-based and structure-based methods to screen over 13,5 million possible cross-reactive peptide pairs from BCG and SARS-CoV-2. Our study produced (i) a list of immunogenic BCG-derived peptides that may prime T cell cross-reactivity against SARS-CoV-2, (ii) a large dataset of modeled peptide-HLA structures for the screened targets, and (iii) new computational methods for structure-based screenings that can be used by others in future studies. Our study expands the list of BCG peptides potentially involved in T cell cross-reactivity with SARS-CoV-2-derived peptides, and identifies multiple high-density "neighborhoods" of cross-reactive peptides which could be driving heterologous immunity induced by BCG vaccination, therefore providing insights for future vaccine development efforts. - R. Fasoulis, G. Paliouras, and L. E. Kavraki, “Graph representation learning for structural proteomics,” Emerging Topics in Life Sciences, Oct. 2021.
BibTeX
@article{fasoulis2021-grlsp, title = {Graph representation learning for structural proteomics}, author = {Fasoulis, Romanos and Paliouras, Georgios and Kavraki, Lydia E.}, journal = {Emerging Topics in Life Sciences}, month = oct, year = {2021}, doi = {10.1042/ETLS20210225}, abstract = {The field of structural proteomics, which is focused on studying the structure–function relationship of proteins and protein complexes, is experiencing rapid growth. Since the early 2000s, structural databases such as the Protein Data Bank are storing increasing amounts of protein structural data, in addition to modeled structures becoming increasingly available. This, combined with the recent advances in graph-based machine-learning models, enables the use of protein structural data in predictive models, with the goal of creating tools that will advance our understanding of protein function. Similar to using graph learning tools to molecular graphs, which currently undergo rapid development, there is also an increasing trend in using graph learning approaches on protein structures. In this short review paper, we survey studies that use graph learning techniques on proteins, and examine their successes and shortcomings, while also discussing future directions.}, issn = {2397-8554}, url = {https://doi.org/10.1042/ETLS20210225} }DetailsAbstract
The field of structural proteomics, which is focused on studying the structure–function relationship of proteins and protein complexes, is experiencing rapid growth. Since the early 2000s, structural databases such as the Protein Data Bank are storing increasing amounts of protein structural data, in addition to modeled structures becoming increasingly available. This, combined with the recent advances in graph-based machine-learning models, enables the use of protein structural data in predictive models, with the goal of creating tools that will advance our understanding of protein function. Similar to using graph learning tools to molecular graphs, which currently undergo rapid development, there is also an increasing trend in using graph learning approaches on protein structures. In this short review paper, we survey studies that use graph learning techniques on proteins, and examine their successes and shortcomings, while also discussing future directions. - E. E. Litsa, P. Das, and L. E. Kavraki, “Machine learning models in the prediction of drug metabolism: challenges and future
perspectives,” Expert Opinion on Drug Metabolism & Toxicology, vol. 0, no. 0, pp. 1–3, 2021. PMID: 34706606
BibTeX
@article{litsa2021-expert-opinion, title = {Machine learning models in the prediction of drug metabolism: challenges and future perspectives}, author = {Litsa, Eleni E. and Das, Payel and Kavraki, Lydia E.}, journal = {Expert Opinion on Drug Metabolism \& Toxicology}, year = {2021}, volume = {0}, number = {0}, pages = {1--3}, doi = {10.1080/17425255.2021.1998454}, abstract = {Metabolism can be the underlying cause of drug adverse effects and diminished efficacy. Metabolic reactions in the human body, mediated mainly by enzymes, may transform the administered drug into metabolites that exhibit different biological activity. As a general rule, metabolic reactions deactivate a drug; however, off-target effects or toxicity, resulting from the formed metabolites, cannot be excluded. On the flip side, metabolism is necessary for the formation of the active substance in the case of prodrugs. In scenarios where multiple drugs are co-administered, the presence of a drug may inhibit or further induce the clearance of another setting metabolism as one of the underlying causes of drug–drug interactions. As a result, the metabolic fate of a candidate drug needs to be thoroughly investigated during the drug development process.}, note = {PMID: 34706606}, publisher = {Taylor \& Francis}, url = {https://doi.org/10.1080/17425255.2021.1998454} }DetailsAbstract
Metabolism can be the underlying cause of drug adverse effects and diminished efficacy. Metabolic reactions in the human body, mediated mainly by enzymes, may transform the administered drug into metabolites that exhibit different biological activity. As a general rule, metabolic reactions deactivate a drug; however, off-target effects or toxicity, resulting from the formed metabolites, cannot be excluded. On the flip side, metabolism is necessary for the formation of the active substance in the case of prodrugs. In scenarios where multiple drugs are co-administered, the presence of a drug may inhibit or further induce the clearance of another setting metabolism as one of the underlying causes of drug–drug interactions. As a result, the metabolic fate of a candidate drug needs to be thoroughly investigated during the drug development process. - S. Hall-Swan, D. Devaurs, M. M. Rigo, D. A. Antunes, L. E. Kavraki, and G. Zanatta, “DINC-COVID: A webserver for ensemble docking with flexible SARS-CoV-2 proteins,” Computers in Biology and Medicine, vol. 139, p. 104943, 2021.
BibTeX
@article{hall-swan2021-dinc-covid, title = {DINC-COVID: A webserver for ensemble docking with flexible SARS-CoV-2 proteins}, author = {Hall-Swan, Sarah and Devaurs, Didier and Rigo, Mauricio M. and Antunes, Dinler A. and Kavraki, Lydia E. and Zanatta, Geancarlo}, journal = {Computers in Biology and Medicine}, year = {2021}, volume = {139}, pages = {104943}, doi = {https://doi.org/10.1016/j.compbiomed.2021.104943}, abstract = {An unprecedented research effort has been undertaken in response to the ongoing COVID-19 pandemic. This has included the determination of hundreds of crystallographic structures of SARS-CoV-2 proteins, and numerous virtual screening projects searching large compound libraries for potential drug inhibitors. Unfortunately, these initiatives have had very limited success in producing effective inhibitors against SARS-CoV-2 proteins. A reason might be an often overlooked factor in these computational efforts: receptor flexibility. To address this issue we have implemented a computational tool for ensemble docking with SARS-CoV-2 proteins. We have extracted representative ensembles of protein conformations from the Protein Data Bank and from in silico molecular dynamics simulations. Twelve pre-computed ensembles of SARS-CoV-2 protein conformations have now been made available for ensemble docking via a user-friendly webserver called DINC-COVID (dinc-covid.kavrakilab.org). We have validated DINC-COVID using data on tested inhibitors of two SARS-CoV-2 proteins, obtaining good correlations between docking-derived binding energies and experimentally-determined binding affinities. Some of the best results have been obtained on a dataset of large ligands resolved via room temperature crystallography, and therefore capturing alternative receptor conformations. In addition, we have shown that the ensembles available in DINC-COVID capture different ranges of receptor flexibility, and that this diversity is useful in finding alternative binding modes of ligands. Overall, our work highlights the importance of accounting for receptor flexibility in docking studies, and provides a platform for the identification of new inhibitors against SARS-CoV-2 proteins.}, issn = {0010-4825}, keyword = {COVID-19, SARS-CoV-2, Molecular docking, Ensemble docking, Receptor flexibility, Molecular dynamics}, url = {https://www.sciencedirect.com/science/article/pii/S001048252100737X} }DetailsAbstract
An unprecedented research effort has been undertaken in response to the ongoing COVID-19 pandemic. This has included the determination of hundreds of crystallographic structures of SARS-CoV-2 proteins, and numerous virtual screening projects searching large compound libraries for potential drug inhibitors. Unfortunately, these initiatives have had very limited success in producing effective inhibitors against SARS-CoV-2 proteins. A reason might be an often overlooked factor in these computational efforts: receptor flexibility. To address this issue we have implemented a computational tool for ensemble docking with SARS-CoV-2 proteins. We have extracted representative ensembles of protein conformations from the Protein Data Bank and from in silico molecular dynamics simulations. Twelve pre-computed ensembles of SARS-CoV-2 protein conformations have now been made available for ensemble docking via a user-friendly webserver called DINC-COVID (dinc-covid.kavrakilab.org). We have validated DINC-COVID using data on tested inhibitors of two SARS-CoV-2 proteins, obtaining good correlations between docking-derived binding energies and experimentally-determined binding affinities. Some of the best results have been obtained on a dataset of large ligands resolved via room temperature crystallography, and therefore capturing alternative receptor conformations. In addition, we have shown that the ensembles available in DINC-COVID capture different ranges of receptor flexibility, and that this diversity is useful in finding alternative binding modes of ligands. Overall, our work highlights the importance of accounting for receptor flexibility in docking studies, and provides a platform for the identification of new inhibitors against SARS-CoV-2 proteins.