Structural modeling of peptide-HLA complexes presenting a melanoma-associated antigen for cross-reactivity assessment
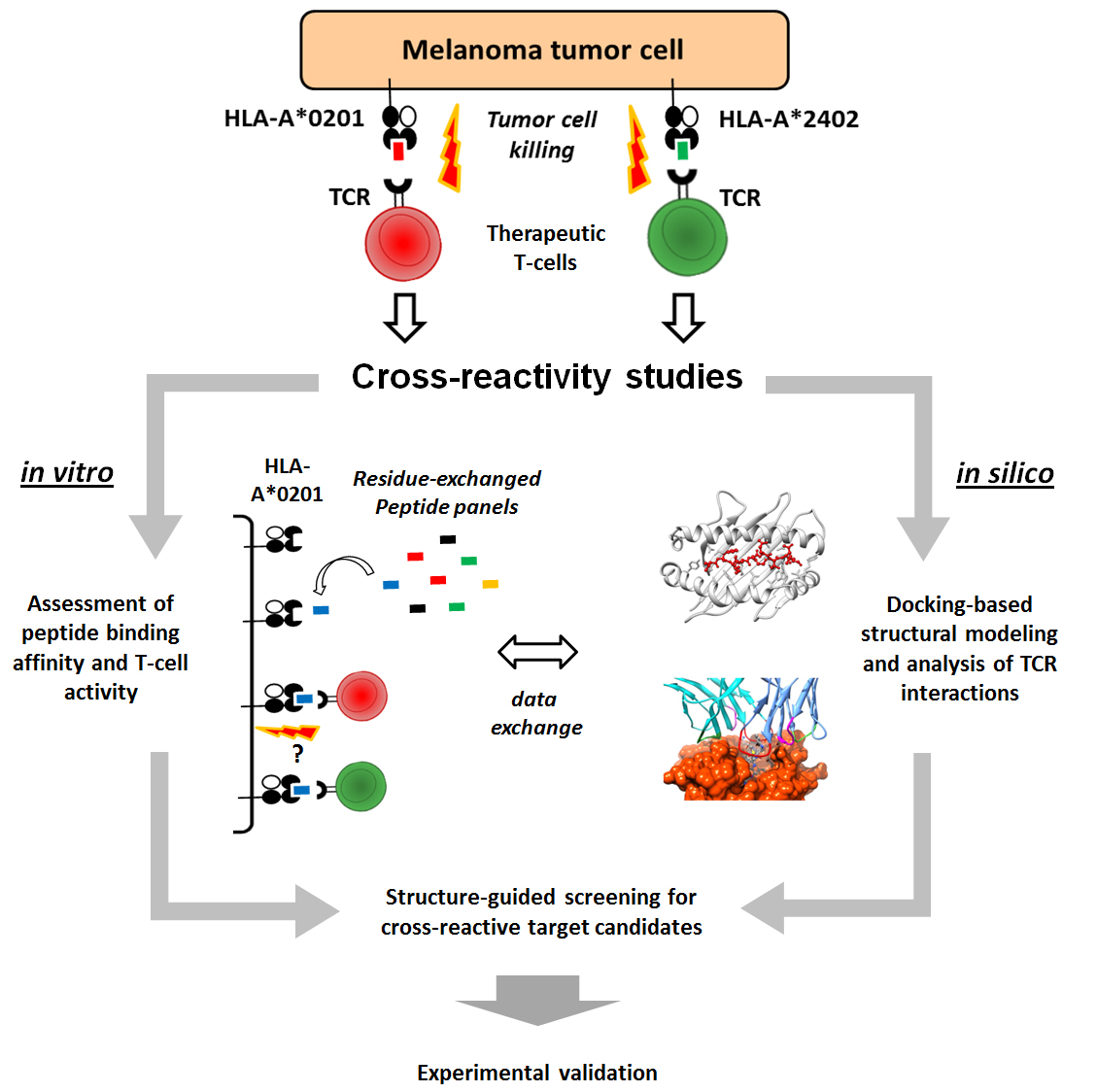
Immunotherapy has become one of the most active fields in cancer research, focusing on treatments that make use of properties of the immune system to specifically recognize and eliminate tumor cells. The so-called "killer" T-cell lymphocytes, a particular type of white blood cell, are of special interest in this context. These T-cells are naturally involved in the surveillance against diseased and infected cells. More specifically, a tumor cell will display at its surface certain tumor-derived peptides (i.e., fragments of proteins). In turn, these peptides will serve as molecular "flags" that can be recognized by the T-cells. This recognition triggers toxic mechanisms within the T-cell that lead to the killing of the tumor cell. T-cell-based immunotherapies have proven to be very effective at eradicating tumor cells in many cancer patients, but its application in a larger scale have been prevented by safety concerns. For instance, at least 5 deaths were reported in recent clinical trials, being associated to unexpected off-target toxicity of the therapeutic T-cells in the heart and the brain. Predicting these T-cell cross-reactivities is a very challenging task, given the complexity of this immunological phenomena. Here we propose an innovative approach to perform T-cell cross-reactivity assessment. We will combine diverse computational methods with standard experimental assays in order to define T-cell-specific motifs that can be used for large-scale screening of potentially dangerous cross-reactive peptide-targets expressed by normal cells. We will apply these methods in the context of two newly-identified melanoma-associated peptides, using our new methods to select safe T-cell lines that can then be used in clinical trials. By developing these new methods for cross-reactivity assessment we will contribute to the development of other T-cell-based therapies, and open new research avenues that will have a positive impact in other human diseases.
This work has been supported by grant CPRIT RP170508.
Related Publications
- K. R. Jackson, D. A. Antunes, A. H. Talukder, A. R. Maleki, K. Amagai, A. Salmon, A. S. Katailiha, Y. Chiu, R. Fasoulis, M. M. Rigo, J. R. Abella, B. D. Melendez, F. Li, Y. Sun, H. M. Sonnemann, V. Belousov, F. Frenkel, S. Justesen, A. Makaju, Y. Liu, D. Horn, D. Lopez-Ferrer, A. F. Huhmer, P. Hwu, J. Roszik, D. Hawke, L. E. Kavraki, and G. Lizée, “Charge-based interactions through peptide position 4 drive diversity of antigen
presentation by human leukocyte antigen class I molecules,” PNAS Nexus, vol. 1, no. 3, Aug. 2022.
BibTeX
@article{jackson2022-charge-interactions, author = {Jackson, Kyle R and Antunes, Dinler A and Talukder, Amjad H and Maleki, Ariana R and Amagai, Kano and Salmon, Avery and Katailiha, Arjun S and Chiu, Yulun and Fasoulis, Romanos and Rigo, Maurício Menegatti and Abella, Jayvee R and Melendez, Brenda D and Li, Fenge and Sun, Yimo and Sonnemann, Heather M and Belousov, Vladislav and Frenkel, Felix and Justesen, Sune and Makaju, Aman and Liu, Yang and Horn, David and Lopez-Ferrer, Daniel and Huhmer, Andreas F and Hwu, Patrick and Roszik, Jason and Hawke, David and Kavraki, Lydia E and Lizée, Gregory}, title = {{Charge-based interactions through peptide position 4 drive diversity of antigen presentation by human leukocyte antigen class I molecules}}, journal = {PNAS Nexus}, volume = {1}, number = {3}, year = {2022}, month = aug, abstract = {Human leukocyte antigen class I (HLA-I) molecules bind and present peptides at the cell surface to facilitate the induction of appropriate CD8+ T cell-mediated immune responses to pathogen- and self-derived proteins. The HLA-I peptide-binding cleft contains dominant anchor sites in the B and F pockets that interact primarily with amino acids at peptide position 2 and the C-terminus, respectively. Nonpocket peptide–HLA interactions also contribute to peptide binding and stability, but these secondary interactions are thought to be unique to individual HLA allotypes or to specific peptide antigens. Here, we show that two positively charged residues located near the top of peptide-binding cleft facilitate interactions with negatively charged residues at position 4 of presented peptides, which occur at elevated frequencies across most HLA-I allotypes. Loss of these interactions was shown to impair HLA-I/peptide binding and complex stability, as demonstrated by both in vitro and in silico experiments. Furthermore, mutation of these Arginine-65 (R65) and/or Lysine-66 (K66) residues in HLA-A*02:01 and A*24:02 significantly reduced HLA-I cell surface expression while also reducing the diversity of the presented peptide repertoire by up to 5-fold. The impact of the R65 mutation demonstrates that nonpocket HLA-I/peptide interactions can constitute anchor motifs that exert an unexpectedly broad influence on HLA-I-mediated antigen presentation. These findings provide fundamental insights into peptide antigen binding that could broadly inform epitope discovery in the context of viral vaccine development and cancer immunotherapy.}, issn = {2752-6542}, doi = {10.1093/pnasnexus/pgac124}, url = {https://doi.org/10.1093/pnasnexus/pgac124} }DetailsAbstract
Human leukocyte antigen class I (HLA-I) molecules bind and present peptides at the cell surface to facilitate the induction of appropriate CD8+ T cell-mediated immune responses to pathogen- and self-derived proteins. The HLA-I peptide-binding cleft contains dominant anchor sites in the B and F pockets that interact primarily with amino acids at peptide position 2 and the C-terminus, respectively. Nonpocket peptide–HLA interactions also contribute to peptide binding and stability, but these secondary interactions are thought to be unique to individual HLA allotypes or to specific peptide antigens. Here, we show that two positively charged residues located near the top of peptide-binding cleft facilitate interactions with negatively charged residues at position 4 of presented peptides, which occur at elevated frequencies across most HLA-I allotypes. Loss of these interactions was shown to impair HLA-I/peptide binding and complex stability, as demonstrated by both in vitro and in silico experiments. Furthermore, mutation of these Arginine-65 (R65) and/or Lysine-66 (K66) residues in HLA-A*02:01 and A*24:02 significantly reduced HLA-I cell surface expression while also reducing the diversity of the presented peptide repertoire by up to 5-fold. The impact of the R65 mutation demonstrates that nonpocket HLA-I/peptide interactions can constitute anchor motifs that exert an unexpectedly broad influence on HLA-I-mediated antigen presentation. These findings provide fundamental insights into peptide antigen binding that could broadly inform epitope discovery in the context of viral vaccine development and cancer immunotherapy. - E. E. Litsa, P. Das, and L. E. Kavraki, “Prediction of drug metabolites using neural machine translation,” Chemical Science, no. 11, pp. 12777–12788, Sep. 2020.
BibTeX
@article{litsa2020-metabolite-prediction, title = {Prediction of drug metabolites using neural machine translation}, author = {Litsa, Eleni E. and Das, Payel and Kavraki, Lydia E.}, journal = {Chemical Science}, month = sep, year = {2020}, pages = {12777--12788}, doi = {10.1039/D0SC02639E}, abstract = {Metabolic processes in the human body can alter the structure of a drug affecting its efficacy and safety. As a result, the investigation of the metabolic fate of a candidate drug is an essential part of drug design studies. Computational approaches have been developed for the prediction of possible drug metabolites in an effort to assist the traditional and resource-demanding experimental route. Current methodologies are based upon metabolic transformation rules, which are tied to specific enzyme families and therefore lack generalization, and additionally may involve manual work from experts limiting scalability. We present a rule-free, end-to-end learning-based method for predicting possible human metabolites of small molecules including drugs. The metabolite prediction task is approached as a sequence translation problem with chemical compounds represented using the SMILES notation. We perform transfer learning on a deep learning transformer model for sequence translation, originally trained on chemical reaction data, to predict the outcome of human metabolic reactions. We further build an ensemble model to account for multiple and diverse metabolites. Extensive evaluation reveals that the proposed method generalizes well to different enzyme families, as it can correctly predict metabolites through phase I and phase II drug metabolism as well as other enzymes. Compared to existing rule-based approaches, our method has equivalent performance on the major enzyme families while it additionally finds metabolites through less common enzymes. Our results indicate that the proposed approach can provide a comprehensive study of drug metabolism that does not restrict to the major enzyme families and does not require the extraction of transformation rules.}, issue = {11}, publisher = {The Royal Society of Chemistry}, url = {http://dx.doi.org/10.1039/D0SC02639E} }DetailsAbstract
Metabolic processes in the human body can alter the structure of a drug affecting its efficacy and safety. As a result, the investigation of the metabolic fate of a candidate drug is an essential part of drug design studies. Computational approaches have been developed for the prediction of possible drug metabolites in an effort to assist the traditional and resource-demanding experimental route. Current methodologies are based upon metabolic transformation rules, which are tied to specific enzyme families and therefore lack generalization, and additionally may involve manual work from experts limiting scalability. We present a rule-free, end-to-end learning-based method for predicting possible human metabolites of small molecules including drugs. The metabolite prediction task is approached as a sequence translation problem with chemical compounds represented using the SMILES notation. We perform transfer learning on a deep learning transformer model for sequence translation, originally trained on chemical reaction data, to predict the outcome of human metabolic reactions. We further build an ensemble model to account for multiple and diverse metabolites. Extensive evaluation reveals that the proposed method generalizes well to different enzyme families, as it can correctly predict metabolites through phase I and phase II drug metabolism as well as other enzymes. Compared to existing rule-based approaches, our method has equivalent performance on the major enzyme families while it additionally finds metabolites through less common enzymes. Our results indicate that the proposed approach can provide a comprehensive study of drug metabolism that does not restrict to the major enzyme families and does not require the extraction of transformation rules. - D. A. Antunes, J. R. Abella, S. Hall-Swan, D. Devaurs, A. Conev, M. Moll, G. Lizée, and L. E. Kavraki, “HLA-Arena: a customizable environment for the structural modeling and analysis of
peptide-HLA complexes for cancer immunotherapy,” JCO Clinical Cancer Informatics, vol. 4, pp. 623–636, Jul. 2020. PMID: 32667823, PMCID: 7397777
BibTeX
@article{antunes2020-hla-arena, title = {{HLA}-{A}rena: a customizable environment for the structural modeling and analysis of peptide-{HLA} complexes for cancer immunotherapy}, author = {Antunes, Dinler A. and Abella, Jayvee R. and Hall-Swan, Sarah and Devaurs, Didier and Conev, Anja and Moll, Mark and Liz\'{e}e, Gregory and Kavraki, Lydia E.}, journal = {JCO Clinical Cancer Informatics}, month = jul, year = {2020}, volume = {4}, pages = {623--636}, doi = {10.1200/CCI.19.00123}, abstract = {PURPOSE: HLA protein receptors play a key role in cellular immunity. They bind intracellular peptides and display them for recognition by T-cell lymphocytes. Because T-cell activation is partially driven by structural features of these peptide-HLA complexes, their structural modeling and analysis are becoming central components of cancer immunotherapy projects. Unfortunately, this kind of analysis is limited by the small number of experimentally determined structures of peptide-HLA complexes. Overcoming this limitation requires developing novel computational methods to model and analyze peptide-HLA structures. METHODS: Here we describe a new platform for the structural modeling and analysis of peptide-HLA complexes, called HLA-Arena, which we have implemented using Jupyter Notebook and Docker. It is a customizable environment that facilitates the use of computational tools, such as APE-Gen and DINC, which we have previously applied to peptide-HLA complexes. By integrating other commonly used tools, such as MODELLER and MHCflurry, this environment includes support for diverse tasks in structural modeling, analysis, and visualization. RESULTS: To illustrate the capabilities of HLA-Arena, we describe 3 example workflows applied to peptide-HLA complexes. Leveraging the strengths of our tools, DINC and APE-Gen, the first 2 workflows show how to perform geometry prediction for peptide-HLA complexes and structure-based binding prediction, respectively. The third workflow presents an example of large-scale virtual screening of peptides for multiple HLA alleles. CONCLUSION: These workflows illustrate the potential benefits of HLA-Arena for the structural modeling and analysis of peptide-HLA complexes. Because HLA-Arena can easily be integrated within larger computational pipelines, we expect its potential impact to vastly increase. For instance, it could be used to conduct structural analyses for personalized cancer immunotherapy, neoantigen discovery, or vaccine development.}, keyword = {fundamentals of protein modeling, proteins and drugs, other biomedical computing}, note = {PMID: 32667823, PMCID: 7397777} }DetailsAbstract
PURPOSE: HLA protein receptors play a key role in cellular immunity. They bind intracellular peptides and display them for recognition by T-cell lymphocytes. Because T-cell activation is partially driven by structural features of these peptide-HLA complexes, their structural modeling and analysis are becoming central components of cancer immunotherapy projects. Unfortunately, this kind of analysis is limited by the small number of experimentally determined structures of peptide-HLA complexes. Overcoming this limitation requires developing novel computational methods to model and analyze peptide-HLA structures. METHODS: Here we describe a new platform for the structural modeling and analysis of peptide-HLA complexes, called HLA-Arena, which we have implemented using Jupyter Notebook and Docker. It is a customizable environment that facilitates the use of computational tools, such as APE-Gen and DINC, which we have previously applied to peptide-HLA complexes. By integrating other commonly used tools, such as MODELLER and MHCflurry, this environment includes support for diverse tasks in structural modeling, analysis, and visualization. RESULTS: To illustrate the capabilities of HLA-Arena, we describe 3 example workflows applied to peptide-HLA complexes. Leveraging the strengths of our tools, DINC and APE-Gen, the first 2 workflows show how to perform geometry prediction for peptide-HLA complexes and structure-based binding prediction, respectively. The third workflow presents an example of large-scale virtual screening of peptides for multiple HLA alleles. CONCLUSION: These workflows illustrate the potential benefits of HLA-Arena for the structural modeling and analysis of peptide-HLA complexes. Because HLA-Arena can easily be integrated within larger computational pipelines, we expect its potential impact to vastly increase. For instance, it could be used to conduct structural analyses for personalized cancer immunotherapy, neoantigen discovery, or vaccine development. - J. R. Abella, D. A. Antunes, C. Clementi, and L. E. Kavraki, “Large-scale structure-based prediction of stable peptide binding to Class I HLAs using
random forests,” Frontiers in Immunology, vol. 11, no. 1583, Jul. 2020. PMID: 32793224, PMCID: PMC7387700
BibTeX
@article{abella2020-frontiers-random-forest, title = {Large-scale structure-based prediction of stable peptide binding to Class I HLAs using random forests}, author = {Abella, Jayvee R. and Antunes, Dinler A. and Clementi, Cecilia and Kavraki, Lydia E.}, journal = {Frontiers in Immunology}, month = jul, year = {2020}, volume = {11}, number = {1583}, doi = {10.3389/fimmu.2020.01583}, abstract = {Prediction of stable peptide binding to Class I HLAs is an important component for designing immunotherapies. While the best performing predictors are based on machine learning algorithms trained on peptide-HLA (pHLA) sequences, the use of structure for training predictors deserves further exploration. Given enough pHLA structures, a predictor based on the residue-residue interactions found in these structures has the potential to generalize for alleles with little or no experimental data. We have previously developed APE-Gen, a modeling approach able to produce pHLA structures in a scalable manner. In this work we use APE-Gen to model over 150,000 pHLA structures, the largest dataset of its kind, which were used to train a structure-based pan-allele model. We extract simple, homogenous features based on residue-residue distances between peptide and HLA, and build a random forest model for predicting stable pHLA binding. Our model achieves competitive AUROC values on leave-one-allele-out validation tests using significantly less data when compared to popular sequence-based methods. Additionally, our model offers an interpretation analysis that can reveal how the model composes the features to arrive at any given prediction. This interpretation analysis can be used to check if the model is in line with chemical intuition, and we showcase particular examples. Our work is a significant step towards using structure to achieve generalizable and more interpretable prediction for stable pHLA binding.}, keyword = {fundamentals of protein modeling, proteins and drugs, other biomedical computing}, note = {PMID: 32793224, PMCID: PMC7387700} }DetailsAbstract
Prediction of stable peptide binding to Class I HLAs is an important component for designing immunotherapies. While the best performing predictors are based on machine learning algorithms trained on peptide-HLA (pHLA) sequences, the use of structure for training predictors deserves further exploration. Given enough pHLA structures, a predictor based on the residue-residue interactions found in these structures has the potential to generalize for alleles with little or no experimental data. We have previously developed APE-Gen, a modeling approach able to produce pHLA structures in a scalable manner. In this work we use APE-Gen to model over 150,000 pHLA structures, the largest dataset of its kind, which were used to train a structure-based pan-allele model. We extract simple, homogenous features based on residue-residue distances between peptide and HLA, and build a random forest model for predicting stable pHLA binding. Our model achieves competitive AUROC values on leave-one-allele-out validation tests using significantly less data when compared to popular sequence-based methods. Additionally, our model offers an interpretation analysis that can reveal how the model composes the features to arrive at any given prediction. This interpretation analysis can be used to check if the model is in line with chemical intuition, and we showcase particular examples. Our work is a significant step towards using structure to achieve generalizable and more interpretable prediction for stable pHLA binding. - T. Arns, D. A. Antunes, J. R. Abella, M. M. Rigo, L. E. Kavraki, S. Giuliatti, and E. A. Donadi, “Structural Modeling and Molecular Dynamics of the Immune Checkpoint Molecule HLA-G,” Frontiers in Immunology, vol. 11, p. 2882, 2020. PMCID: PMC7677236
BibTeX
@article{thais2020, title = {Structural Modeling and Molecular Dynamics of the Immune Checkpoint Molecule HLA-G}, author = {Arns, Thais and Antunes, Dinler A. and Abella, Jayvee R. and Rigo, Maurício M. and Kavraki, Lydia E. and Giuliatti, Silvana and Donadi, Eduardo A.}, journal = {Frontiers in Immunology}, year = {2020}, volume = {11}, pages = {2882}, doi = {10.3389/fimmu.2020.575076}, abstract = {HLA-G is considered to be an immune checkpoint molecule, a function that is closely linked to the structure and dynamics of the different HLA-G isoforms. Unfortunately, little is known about the structure and dynamics of these isoforms. For instance, there are only seven crystal structures of HLA-G molecules, being all related to a single isoform, and in some cases lacking important residues associated to the interaction with leukocyte receptors. In addition, they lack information on the dynamics of both membrane-bound HLA-G forms, and soluble forms. We took advantage of in silico strategies to disclose the dynamic behavior of selected HLA-G forms, including the membrane-bound HLA-G1 molecule, soluble HLA-G1 dimer, and HLA-G5 isoform. Both the membrane-bound HLA-G1 molecule and the soluble HLA-G1 dimer were quite stable. Residues involved in the interaction with ILT2 and ILT4 receptors (α3 domain) were very close to the lipid bilayer in the complete HLA-G1 molecule, which might limit accessibility. On the other hand, these residues can be completely exposed in the soluble HLA-G1 dimer, due to the free rotation of the disulfide bridge (Cys42/Cys42). In fact, we speculate that this free rotation of each protomer (i.e., the chains composing the dimer) could enable alternative binding modes for ILT2/ILT4 receptors, which in turn could be associated with greater affinity of the soluble HLA-G1 dimer. Structural analysis of the HLA-G5 isoform demonstrated higher stability for the complex containing the peptide and coupled β2-microglobulin, while structures lacking such domains were significantly unstable. This study reports for the first time structural conformations for the HLA-G5 isoform and the dynamic behavior of HLA-G1 molecules under simulated biological conditions. All modeled structures were made available through GitHub (https://github.com/KavrakiLab/), enabling their use as templates for modeling other alleles and isoforms, as well as for other computational analyses to investigate key molecular interactions.}, issn = {1664-3224}, note = {PMCID: PMC7677236}, url = {https://www.frontiersin.org/article/10.3389/fimmu.2020.575076} }DetailsAbstract
HLA-G is considered to be an immune checkpoint molecule, a function that is closely linked to the structure and dynamics of the different HLA-G isoforms. Unfortunately, little is known about the structure and dynamics of these isoforms. For instance, there are only seven crystal structures of HLA-G molecules, being all related to a single isoform, and in some cases lacking important residues associated to the interaction with leukocyte receptors. In addition, they lack information on the dynamics of both membrane-bound HLA-G forms, and soluble forms. We took advantage of in silico strategies to disclose the dynamic behavior of selected HLA-G forms, including the membrane-bound HLA-G1 molecule, soluble HLA-G1 dimer, and HLA-G5 isoform. Both the membrane-bound HLA-G1 molecule and the soluble HLA-G1 dimer were quite stable. Residues involved in the interaction with ILT2 and ILT4 receptors (α3 domain) were very close to the lipid bilayer in the complete HLA-G1 molecule, which might limit accessibility. On the other hand, these residues can be completely exposed in the soluble HLA-G1 dimer, due to the free rotation of the disulfide bridge (Cys42/Cys42). In fact, we speculate that this free rotation of each protomer (i.e., the chains composing the dimer) could enable alternative binding modes for ILT2/ILT4 receptors, which in turn could be associated with greater affinity of the soluble HLA-G1 dimer. Structural analysis of the HLA-G5 isoform demonstrated higher stability for the complex containing the peptide and coupled β2-microglobulin, while structures lacking such domains were significantly unstable. This study reports for the first time structural conformations for the HLA-G5 isoform and the dynamic behavior of HLA-G1 molecules under simulated biological conditions. All modeled structures were made available through GitHub (https://github.com/KavrakiLab/), enabling their use as templates for modeling other alleles and isoforms, as well as for other computational analyses to investigate key molecular interactions. - J. R. Abella, D. Antunes, K. Jackson, G. Lizée, C. Clementi, and L. E. Kavraki, “Markov state modeling reveals alternative unbinding pathways for
peptide–MHC complexes,” Proceedings of the National Academy of Sciences, vol. 117, no. 48, pp. 30610–30618, 2020.
BibTeX
@article{abella2020-pnas, title = {Markov state modeling reveals alternative unbinding pathways for peptide{\textendash}{MHC} complexes}, author = {Abella, Jayvee R. and Antunes, Dinler and Jackson, Kyle and Liz{\'e}e, Gregory and Clementi, Cecilia and Kavraki, Lydia E.}, journal = {Proceedings of the National Academy of Sciences}, year = {2020}, volume = {117}, number = {48}, pages = {30610--30618}, doi = {10.1073/pnas.2007246117}, abstract = {Peptide binding to MHC receptors is part of a central biological process that enables our immune system to attack diseased cells. We use molecular simulations to illuminate the mechanisms driving stable peptide{\textendash}MHC binding. Our simulation framework produces an atomistic model of the unbinding dynamics for a given peptide{\textendash}MHC, which quantifies transitions between the major states of the system (bound, intermediate, and unbound). We applied this framework to study the binding of a SARS-CoV peptide to the HLA-A*24:02 receptor. This work revealed the unexpected importance of peptide{\textquoteright}s position 4 in driving the stability of the complex, a finding with broader biomedical implications. Our methods can be applied to other peptide{\textendash}MHC complexes, requiring only a 3D model as input.Peptide binding to major histocompatibility complexes (MHCs) is a central component of the immune system, and understanding the mechanism behind stable peptide{\textendash}MHC binding will aid the development of immunotherapies. While MHC binding is mostly influenced by the identity of the so-called anchor positions of the peptide, secondary interactions from nonanchor positions are known to play a role in complex stability. However, current MHC-binding prediction methods lack an analysis of the major conformational states and might underestimate the impact of secondary interactions. In this work, we present an atomically detailed analysis of peptide{\textendash}MHC binding that can reveal the contributions of any interaction toward stability. We propose a simulation framework that uses both umbrella sampling and adaptive sampling to generate a Markov state model (MSM) for a coronavirus-derived peptide (QFKDNVILL), bound to one of the most prevalent MHC receptors in humans (HLA-A24:02). While our model reaffirms the importance of the anchor positions of the peptide in establishing stable interactions, our model also reveals the underestimated importance of position 4 (p4), a nonanchor position. We confirmed our results by simulating the impact of specific peptide mutations and validated these predictions through competitive binding assays. By comparing the MSM of the wild-type system with those of the D4A and D4P mutations, our modeling reveals stark differences in unbinding pathways. The analysis presented here can be applied to any peptide{\textendash}MHC complex of interest with a structural model as input, representing an important step toward comprehensive modeling of the MHC class I pathway.Code for umbrella sampling, adaptive sampling, and MSM analysis, as well as representative structures, can be found in Github at https://github.com/KavrakiLab/adaptive-samplingpmhc. Simulation data are available upon request.}, issn = {0027-8424}, publisher = {National Academy of Sciences}, url = {https://www.pnas.org/content/117/48/30610} }DetailsAbstract
Peptide binding to MHC receptors is part of a central biological process that enables our immune system to attack diseased cells. We use molecular simulations to illuminate the mechanisms driving stable peptide–MHC binding. Our simulation framework produces an atomistic model of the unbinding dynamics for a given peptide–MHC, which quantifies transitions between the major states of the system (bound, intermediate, and unbound). We applied this framework to study the binding of a SARS-CoV peptide to the HLA-A*24:02 receptor. This work revealed the unexpected importance of peptide’s position 4 in driving the stability of the complex, a finding with broader biomedical implications. Our methods can be applied to other peptide–MHC complexes, requiring only a 3D model as input.Peptide binding to major histocompatibility complexes (MHCs) is a central component of the immune system, and understanding the mechanism behind stable peptide–MHC binding will aid the development of immunotherapies. While MHC binding is mostly influenced by the identity of the so-called anchor positions of the peptide, secondary interactions from nonanchor positions are known to play a role in complex stability. However, current MHC-binding prediction methods lack an analysis of the major conformational states and might underestimate the impact of secondary interactions. In this work, we present an atomically detailed analysis of peptide–MHC binding that can reveal the contributions of any interaction toward stability. We propose a simulation framework that uses both umbrella sampling and adaptive sampling to generate a Markov state model (MSM) for a coronavirus-derived peptide (QFKDNVILL), bound to one of the most prevalent MHC receptors in humans (HLA-A24:02). While our model reaffirms the importance of the anchor positions of the peptide in establishing stable interactions, our model also reveals the underestimated importance of position 4 (p4), a nonanchor position. We confirmed our results by simulating the impact of specific peptide mutations and validated these predictions through competitive binding assays. By comparing the MSM of the wild-type system with those of the D4A and D4P mutations, our modeling reveals stark differences in unbinding pathways. The analysis presented here can be applied to any peptide–MHC complex of interest with a structural model as input, representing an important step toward comprehensive modeling of the MHC class I pathway.Code for umbrella sampling, adaptive sampling, and MSM analysis, as well as representative structures, can be found in Github at https://github.com/KavrakiLab/adaptive-samplingpmhc. Simulation data are available upon request. - D. Devaurs, D. A. Antunes, S. Hall-Swan, N. Mitchell, M. Moll, G. Lizée, and L. E. Kavraki, “Using parallelized incremental meta-docking can solve the conformational sampling issue
when docking large ligands to proteins,” BMC Molecular and Cell Biology, vol. 20, no. 1, p. 42, Sep. 2019. PMID: 31488048, PMCID: PMC6729087
BibTeX
@article{devaurs2019using-parallelized-incremental-meta-docking, title = {Using parallelized incremental meta-docking can solve the conformational sampling issue when docking large ligands to proteins}, author = {Devaurs, Didier and Antunes, Dinler A and Hall-Swan, Sarah and Mitchell, Nicole and Moll, Mark and Liz{\'e}e, Gregory and Kavraki, Lydia E}, journal = {BMC Molecular and Cell Biology}, month = sep, year = {2019}, volume = {20}, number = {1}, pages = {42}, doi = {10.1186/s12860-019-0218-z}, abstract = {Background: Docking large ligands, and especially peptides, to protein receptors is still considered a challenge in computational structural biology. Besides the issue of accurately scoring the binding modes of a protein-ligand complex produced by a molecular docking tool, the conformational sampling of a large ligand is also often considered a challenge because of its underlying combinatorial complexity. In this study, we evaluate the impact of using parallelized and incremental paradigms on the accuracy and performance of conformational sampling when docking large ligands. We use five datasets of protein-ligand complexes involving ligands that could not be accurately docked by classical protein-ligand docking tools in previous similar studies. Results: Our computational evaluation shows that simply increasing the amount of conformational sampling performed by a protein-ligand docking tool, such as Vina, by running it for longer is rarely beneficial. Instead, it is more efficient and advantageous to run several short instances of this docking tool in parallel and group their results together, in a straightforward parallelized docking protocol. Even greater accuracy and efficiency are achieved by our parallelized incremental meta-docking tool, DINC, showing the additional benefits of its incremental paradigm. Using DINC, we could accurately reproduce the vast majority of the protein-ligand complexes we considered. Conclusions: Our study suggests that, even when trying to dock large ligands to proteins, the conformational sampling of the ligand should no longer be considered an issue, as simple docking protocols using existing tools can solve it. Therefore, scoring should currently be regarded as the biggest unmet challenge in molecular docking. Keywords: molecular docking; protein-ligand docking; protein-peptide docking; conformational sampling; scoring; parallelism; incremental protocol}, keyword = {proteins and drugs}, note = {PMID: 31488048, PMCID: PMC6729087} }DetailsAbstract
Background: Docking large ligands, and especially peptides, to protein receptors is still considered a challenge in computational structural biology. Besides the issue of accurately scoring the binding modes of a protein-ligand complex produced by a molecular docking tool, the conformational sampling of a large ligand is also often considered a challenge because of its underlying combinatorial complexity. In this study, we evaluate the impact of using parallelized and incremental paradigms on the accuracy and performance of conformational sampling when docking large ligands. We use five datasets of protein-ligand complexes involving ligands that could not be accurately docked by classical protein-ligand docking tools in previous similar studies. Results: Our computational evaluation shows that simply increasing the amount of conformational sampling performed by a protein-ligand docking tool, such as Vina, by running it for longer is rarely beneficial. Instead, it is more efficient and advantageous to run several short instances of this docking tool in parallel and group their results together, in a straightforward parallelized docking protocol. Even greater accuracy and efficiency are achieved by our parallelized incremental meta-docking tool, DINC, showing the additional benefits of its incremental paradigm. Using DINC, we could accurately reproduce the vast majority of the protein-ligand complexes we considered. Conclusions: Our study suggests that, even when trying to dock large ligands to proteins, the conformational sampling of the ligand should no longer be considered an issue, as simple docking protocols using existing tools can solve it. Therefore, scoring should currently be regarded as the biggest unmet challenge in molecular docking. Keywords: molecular docking; protein-ligand docking; protein-peptide docking; conformational sampling; scoring; parallelism; incremental protocol - D. A. Antunes, J. R. Abella, D. Devaurs, M. M. Rigo, and L. E. Kavraki, “Structure-based methods for binding mode and binding affinity prediction for
peptide-MHC complexes,” Current Topics in Medicinal Chemistry, vol. 19, no. 1, 2019. PMID: 30582480, PMCID: PMC6361695
BibTeX
@article{antunes2019structure-based-methods-for-binding, title = {Structure-based methods for binding mode and binding affinity prediction for peptide-{MHC} complexes}, author = {Antunes, Dinler A. and Abella, Jayvee R. and Devaurs, Didier and Rigo, Maur\'{i}cio M. and Kavraki, Lydia E.}, journal = {Current Topics in Medicinal Chemistry}, year = {2019}, volume = {19}, number = {1}, doi = {10.2174/1568026619666181224101744}, abstract = {Understanding the mechanisms involved in the activation of an immune response is essential to many fields in human health, including vaccine development and personalized cancer immunotherapy. A central step in the activation of the adaptive immune response is the recognition, by T-cell lymphocytes, of peptides displayed by a special type of receptor known as Major Histocompatibility Complex (MHC). Considering the key role of MHC receptors in T-cell activation, the computational prediction of peptide binding to MHC has been an important goal for many immunological applications. Sequence-based methods have become the gold standard for peptide-MHC binding affinity prediction, but structure-based methods are expected to provide more general predictions (i.e., predictions applicable to all types of MHC receptors). In addition, structural modeling of peptide-MHC complexes has the potential to uncover yet unknown drivers of T-cell activation, thus allowing for the development of better and safer therapies. In this review, we discuss the use of computational methods for the structural modeling of peptide-MHC complexes (i.e., binding mode prediction) and for the structure-based prediction of binding affinity.}, keyword = {proteins and drugs, molecular docking, binding mode prediction, binding affinity prediction, peptide-MHC complexes, immunogenicity, T-cell activation}, note = {PMID: 30582480, PMCID: PMC6361695} }DetailsAbstract
Understanding the mechanisms involved in the activation of an immune response is essential to many fields in human health, including vaccine development and personalized cancer immunotherapy. A central step in the activation of the adaptive immune response is the recognition, by T-cell lymphocytes, of peptides displayed by a special type of receptor known as Major Histocompatibility Complex (MHC). Considering the key role of MHC receptors in T-cell activation, the computational prediction of peptide binding to MHC has been an important goal for many immunological applications. Sequence-based methods have become the gold standard for peptide-MHC binding affinity prediction, but structure-based methods are expected to provide more general predictions (i.e., predictions applicable to all types of MHC receptors). In addition, structural modeling of peptide-MHC complexes has the potential to uncover yet unknown drivers of T-cell activation, thus allowing for the development of better and safer therapies. In this review, we discuss the use of computational methods for the structural modeling of peptide-MHC complexes (i.e., binding mode prediction) and for the structure-based prediction of binding affinity. - J. R. Abella, D. A. Antunes, C. Clementi, and L. E. Kavraki, “APE-Gen: A Fast Method for Generating Ensembles of Bound Peptide-MHC Conformations,” Molecules, vol. 24, no. 5, p. 881, 2019. PMID: 30832312, PMCID: PMC6429480
BibTeX
@article{abella2019-apegen, title = {APE-Gen: A Fast Method for Generating Ensembles of Bound Peptide-MHC Conformations}, author = {Abella, Jayvee R. and Antunes, Dinler A. and Clementi, Cecilia and Kavraki, Lydia E.}, journal = {Molecules}, year = {2019}, volume = {24}, number = {5}, pages = {881}, doi = {10.3390/molecules24050881}, abstract = {The Class I Major Histocompatibility Complex (MHC) is a central protein in immunology as it binds to intracellular peptides and displays them at the cell surface for recognition by T-cells. The structural analysis of bound peptide-MHC complexes (pMHCs) holds the promise of interpretable and general binding prediction (i.e., testing whether a given peptide binds to a given MHC). However, structural analysis is limited in part by the difficulty in modelling pMHCs given the size and flexibility of the peptides that can be presented by MHCs. This article describes APE-Gen (Anchored Peptide-MHC Ensemble Generator), a fast method for generating ensembles of bound pMHC conformations. APE-Gen generates an ensemble of bound conformations by iterated rounds of (i) anchoring the ends of a given peptide near known pockets in the binding site of the MHC, (ii) sampling peptide backbone conformations with loop modelling, and then (iii) performing energy minimization to fix steric clashes, accumulating conformations at each round. APE-Gen takes only minutes on a standard desktop to generate tens of bound conformations, and we show the ability of APE-Gen to sample conformations found in X-ray crystallography even when only sequence information is used as input. APE-Gen has the potential to be useful for its scalability (i.e., modelling thousands of pMHCs or even non-canonical longer peptides) and for its use as a flexible search tool. We demonstrate an example for studying cross-reactivity.}, issn = {1420-3049}, keyword = {fundamentals of protein modeling}, note = {PMID: 30832312, PMCID: PMC6429480}, url = {http://www.mdpi.com/1420-3049/24/5/881} }DetailsAbstract
The Class I Major Histocompatibility Complex (MHC) is a central protein in immunology as it binds to intracellular peptides and displays them at the cell surface for recognition by T-cells. The structural analysis of bound peptide-MHC complexes (pMHCs) holds the promise of interpretable and general binding prediction (i.e., testing whether a given peptide binds to a given MHC). However, structural analysis is limited in part by the difficulty in modelling pMHCs given the size and flexibility of the peptides that can be presented by MHCs. This article describes APE-Gen (Anchored Peptide-MHC Ensemble Generator), a fast method for generating ensembles of bound pMHC conformations. APE-Gen generates an ensemble of bound conformations by iterated rounds of (i) anchoring the ends of a given peptide near known pockets in the binding site of the MHC, (ii) sampling peptide backbone conformations with loop modelling, and then (iii) performing energy minimization to fix steric clashes, accumulating conformations at each round. APE-Gen takes only minutes on a standard desktop to generate tens of bound conformations, and we show the ability of APE-Gen to sample conformations found in X-ray crystallography even when only sequence information is used as input. APE-Gen has the potential to be useful for its scalability (i.e., modelling thousands of pMHCs or even non-canonical longer peptides) and for its use as a flexible search tool. We demonstrate an example for studying cross-reactivity. - D. A. Antunes, M. M. Rigo, M. V. Freitas, M. M. FA, M. Sinigaglia, G. Lizée, L. E. Kavraki, L. K. Selin, M. Cornberg, and G. F. Vieira, “Interpreting T-cell cross-reactivity through structure: implications for TCR-based
cancer immunotherapy,” Front. Immunol., vol. 8, no. 1210, 2017. PMCID: PMC5632759, PMID: 29046675
BibTeX
@article{antunes2017_frontiers-immunol, title = {Interpreting {T}-cell cross-reactivity through structure: implications for {TCR}-based cancer immunotherapy}, author = {Antunes, Dinler A and Rigo, Maur\'{i}cio M and Freitas, Martiela V and FA, Mendes Marcus and Sinigaglia, Marialva and Liz\'{e}e, Gregory and Kavraki, Lydia E and Selin, Liisa K and Cornberg, Markus and Vieira, Gustavo F}, journal = {Front. Immunol.}, year = {2017}, volume = {8}, number = {1210}, doi = {10.3389/fimmu.2017.01210}, abstract = {Immunotherapy has become one of the most promising avenues for cancer treatment, making use of the patient's own immune system to eliminate cancer cells. Clinical trials with T-cell-based immunotherapies have shown dramatic tumor regressions, being effective in multiple cancer types and for many different patients. Unfortunately, this progress was tempered by reports of serious (even fatal) side effects. Such therapies rely on the use of cytotoxic T-cell lymphocytes, an essential part of the adaptive immune system. Cytotoxic T-cells are regularly involved in surveillance and are capable of both eliminating diseased cells and generating protective immunological memory. The specificity of a given T-cell is determined through the structural interaction between the T-cell receptor (TCR) and a peptide-loaded major histocompatibility complex (MHC); i.e., an intracellular peptide–ligand displayed at the cell surface by an MHC molecule. However, a given TCR can recognize different peptide–MHC (pMHC) complexes, which can sometimes trigger an unwanted response that is referred to as T-cell cross-reactivity. This has become a major safety issue in TCR-based immunotherapies, following reports of melanoma-specific T-cells causing cytotoxic damage to healthy tissues (e.g., heart and nervous system). T-cell cross-reactivity has been extensively studied in the context of viral immunology and tissue transplantation. Growing evidence suggests that it is largely driven by structural similarities of seemingly unrelated pMHC complexes. Here, we review recent reports about the existence of pMHC ``hot-spots" for cross-reactivity and propose the existence of a TCR interaction profile (i.e., a refinement of a more general TCR footprint in which some amino acid residues are more important than others in triggering T-cell cross-reactivity). We also make use of available structural data and pMHC models to interpret previously reported cross-reactivity patterns among virus-derived peptides. Our study provides further evidence that structural analyses of pMHC complexes can be used to assess the intrinsic likelihood of cross-reactivity among peptide-targets. Furthermore, we hypothesize that some apparent inconsistencies in reported cross-reactivities, such as a preferential directionality, might also be driven by particular structural features of the targeted pMHC complex. Finally, we explain why TCR-based immunotherapy provides a special context in which meaningful T-cell cross-reactivity predictions can be made.}, keyword = {T-cell cross-reactivity, peptide–MHC complex, cross-reactivity hot-spots, TCR-interacting surface, hierarchical clustering, TCR/pMHC, cancer immunotherapy}, note = {PMCID: PMC5632759, PMID: 29046675} }DetailsAbstract
Immunotherapy has become one of the most promising avenues for cancer treatment, making use of the patient’s own immune system to eliminate cancer cells. Clinical trials with T-cell-based immunotherapies have shown dramatic tumor regressions, being effective in multiple cancer types and for many different patients. Unfortunately, this progress was tempered by reports of serious (even fatal) side effects. Such therapies rely on the use of cytotoxic T-cell lymphocytes, an essential part of the adaptive immune system. Cytotoxic T-cells are regularly involved in surveillance and are capable of both eliminating diseased cells and generating protective immunological memory. The specificity of a given T-cell is determined through the structural interaction between the T-cell receptor (TCR) and a peptide-loaded major histocompatibility complex (MHC); i.e., an intracellular peptide–ligand displayed at the cell surface by an MHC molecule. However, a given TCR can recognize different peptide–MHC (pMHC) complexes, which can sometimes trigger an unwanted response that is referred to as T-cell cross-reactivity. This has become a major safety issue in TCR-based immunotherapies, following reports of melanoma-specific T-cells causing cytotoxic damage to healthy tissues (e.g., heart and nervous system). T-cell cross-reactivity has been extensively studied in the context of viral immunology and tissue transplantation. Growing evidence suggests that it is largely driven by structural similarities of seemingly unrelated pMHC complexes. Here, we review recent reports about the existence of pMHC “hot-spots" for cross-reactivity and propose the existence of a TCR interaction profile (i.e., a refinement of a more general TCR footprint in which some amino acid residues are more important than others in triggering T-cell cross-reactivity). We also make use of available structural data and pMHC models to interpret previously reported cross-reactivity patterns among virus-derived peptides. Our study provides further evidence that structural analyses of pMHC complexes can be used to assess the intrinsic likelihood of cross-reactivity among peptide-targets. Furthermore, we hypothesize that some apparent inconsistencies in reported cross-reactivities, such as a preferential directionality, might also be driven by particular structural features of the targeted pMHC complex. Finally, we explain why TCR-based immunotherapy provides a special context in which meaningful T-cell cross-reactivity predictions can be made.