Structure-based Selection of Tumor-antigens for T-cell Based Immunotherapy
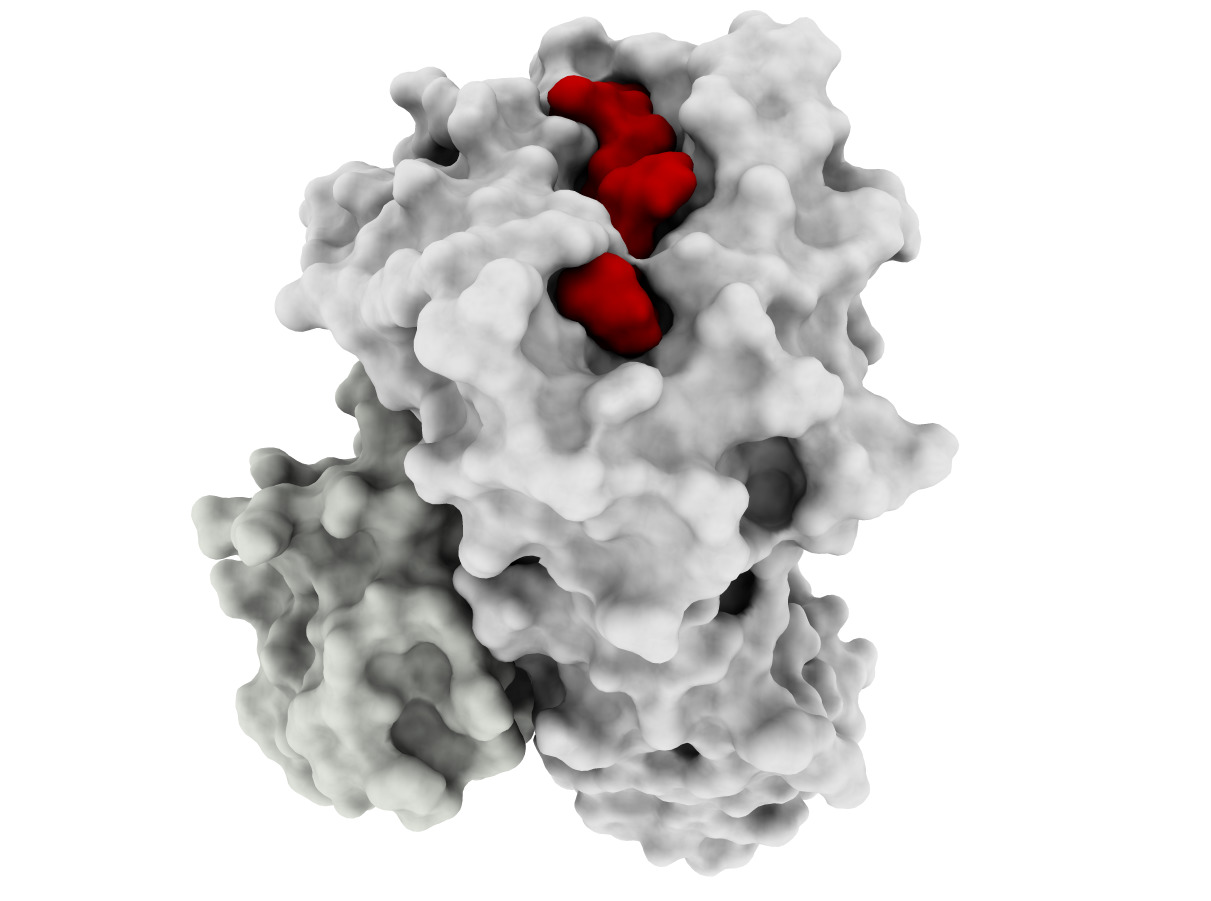
Developing effective cancer treatments remains one of the most important challenges for healthcare, and T-cell based immunotherapy has provided some very positive recent advances in cancer treatment. Cytotoxic T lymphocytes (CTLs) can circulate through the body and are capable of identifying and eliminating tumorigenic cells. The recognition of tumor depends on the specific interaction between the T-cell receptor of CTLs and Human Leucocyte Antigen (HLA) class I molecules at the tumor cell surface, which binds and displays peptides derived from intracellular proteins. Peptide-HLA complexes are presented by all nucleated cells, constituting an efficient surveillance mechanism by which the immune system can recognize aberrant changes within cells of the body. Although CTL surveillance likely evolved to eliminate virally infected cells, this system also provides very promising opportunities for cancer treatment and specifically the development of immune-based therapies. However, such therapies must be highly personalized since most of these tumor-associated peptides are patient-specific. This is due mainly to the high level of HLA diversity within the human population, combined with the fact that each person’s tumor acquires unique genetic aberrations. A further challenge is the identification of tumor-specific peptides that are not also expressed by normal cells, which will likely ensure less off-target effects during therapy. Our long-term goal is to perform structure-guided selection of tumor-derived peptides with potential for immunotherapy, which will also allow structural analysis of different peptide-HLA complexes recognized by a given T-cell; this knowledge will help to prevent dangerous off-target toxicities. The objective of this project is to develop computational tools to enable docking-based modeling of peptide-HLA complexes, starting with HLA variants (allotypes) that are highly prevalent within human population and moving toward others that are less prevalent (for personalized treatment). Our Preliminary Data supports the need for a structural framework to improve the selection of targets for immunotherapy, since current methods have important limitations, particularly with regard to less prevalent HLAs. The central hypothesis is that structure-based analysis can be used to improve peptide target selection for individual HLA allotypes and thus facilitate the development of personalized immunotherapies for all cancer patients. Two specific aims were designed to test this hypothesis. In Specific Aim 1, a docking method will be specifically tailored to make binding predictions of tumor-derived peptides to two highly frequent and well- studied HLAs, HLA-A*2402 and HLA*A1101, collectively expressed by >55% of the world population. In Specific Aim 2, the HLA-A3 superfamily, collectively expressed by >40% of the human population, will be used as a model for extending the methods towards less well-studied HLAs. Innovative computational methods will be applied in this project and cutting-edge experimental resources will be used to train and validate computational methods. The underlying rationale is that developing a computational framework for these prevalent HLA allotypes will facilitate the development of personalized, antigen-specific immunotherapies, which would benefit a much larger number of cancer patients.
This work has been supported by grant NIH 1R21CA209941.
Related Publications
- K. R. Jackson, D. A. Antunes, A. H. Talukder, A. R. Maleki, K. Amagai, A. Salmon, A. S. Katailiha, Y. Chiu, R. Fasoulis, M. M. Rigo, J. R. Abella, B. D. Melendez, F. Li, Y. Sun, H. M. Sonnemann, V. Belousov, F. Frenkel, S. Justesen, A. Makaju, Y. Liu, D. Horn, D. Lopez-Ferrer, A. F. Huhmer, P. Hwu, J. Roszik, D. Hawke, L. E. Kavraki, and G. Lizée, “Charge-based interactions through peptide position 4 drive diversity of antigen
presentation by human leukocyte antigen class I molecules,” PNAS Nexus, vol. 1, no. 3, Aug. 2022.
Details - D. A. Antunes, J. R. Abella, S. Hall-Swan, D. Devaurs, A. Conev, M. Moll, G. Lizée, and L. E. Kavraki, “HLA-Arena: a customizable environment for the structural modeling and analysis of
peptide-HLA complexes for cancer immunotherapy,” JCO Clinical Cancer Informatics, vol. 4, pp. 623–636, Jul. 2020. PMID: 32667823, PMCID: 7397777
Details - D. Devaurs, D. A. Antunes, S. Hall-Swan, N. Mitchell, M. Moll, G. Lizée, and L. E. Kavraki, “Using parallelized incremental meta-docking can solve the conformational sampling issue
when docking large ligands to proteins,” BMC Molecular and Cell Biology, vol. 20, no. 1, p. 42, Sep. 2019. PMID: 31488048, PMCID: PMC6729087
Details - D. A. Antunes, J. R. Abella, D. Devaurs, M. M. Rigo, and L. E. Kavraki, “Structure-based methods for binding mode and binding affinity prediction for
peptide-MHC complexes,” Current Topics in Medicinal Chemistry, vol. 19, no. 1, 2019. PMID: 30582480, PMCID: PMC6361695
Details - D. Devaurs, M. Papanastasiou, D. A. Antunes, J. R. Abella, M. Moll, D. Ricklin, J. D. Lambris, and L. E. Kavraki, “Native state of complement protein C3d analysed via hydrogen exchange and
conformational sampling,” International Journal of Computational Biology and Drug Design, vol. 11, no. 1/2, pp. 90–113, 2018. PMID: 30700993, PMCID: PMC6349257
Details - D. A. Antunes, D. Devaurs, M. Moll, G. Lizée, and L. E. Kavraki, “General prediction of peptide-MHC binding modes using incremental docking: A proof of
concept,” Scientific Reports, vol. 8, p. 4327, 2018. PMCID: PMC5847594, PMID: 29531253
Details - D. A. Antunes, M. Moll, D. Devaurs, K. R. Jackson, G. Lizée, and L. E. Kavraki, “DINC 2.0: a new protein-peptide docking webserver using an incremental approach,” Cancer Research, vol. 77, no. 21, pp. 55–57, Nov. 2017. PMCID: PMC5679007, PMID: 29092940
Details - D. Devaurs, D. A. Antunes, M. Papanastasiou, M. Moll, D. Ricklin, J. D. Lambris, and L. E. Kavraki, “Coarse-grained conformational sampling of protein structure improves the fit to
experimental hydrogen-exchange data,” Frontiers in Molecular Biosciences, vol. 4, no. 13, 2017. PMCID: PMC5344923, PMID: 28344973
Details